Case study
Drug repurposing has become a rich source of safe and effective new therapeutic options against unmet medical needs. With the appearance of SARS CoV2 and the pandemic spread of severe Covid-19, drug repurposing has intensively being used to identify potential new drug candidates. One of the potential drug candidates was a small molecule prodrug with poor solubility in ethanol, practically insoluble in water and rapid hydrolyzation in aqueous media. The drug was marketed as an immediate release tablet for short term acute treatment. Since the drug showed efficacy against viruses including promising results to be effective against SARS CoV2 reformulation was required to achieve sufficient plasma level over the duration of 12 hours for a twice daily oral dosing regimen.
Case study
Drug repurposing has become a rich source of safe and effective new therapeutic options against unmet medical needs. With the appearance of SARS CoV2 and the pandemic spread of severe Covid-19, drug repurposing has intensively being used to identify potential new drug candidates [1]. One of the potential drug candidates was a small molecule prodrug with poor solubility in ethanol, practically insoluble in water and rapid hydrolyzation in aqueous media. The drug was marketed as an immediate release tablet for short term acute treatment. Since the drug showed efficacy against viruses including promising results to be effective against SARS CoV2 reformulation was required to achieve sufficient plasma level over the duration of 12 hours for a twice daily oral dosing regimen. Using available PK data from existing formulations (e.g. IR) combined with application of in-silico tools like PBBK modeling, predictions on formulation enhancements can be made [2]. For this compound it was predicted that an extended release formulation could lower the dose by ~ 40 % while maintaining a constant plasma levels in the therapeutic range. An extended release oral formulation was developed for the clinical trials that meet the release characteristics determined in the Target Product Profile for the new indication. Following successful phase 3 clinical trials of the extended release formulation, the FDA challenged the use of the dissolution test method derived from the immediate release formulation. Due to the BCS classification of the compound and the extended release properties of the formulation a dissolution method should serve as a surrogate for the in-vivo performance with sufficient discriminatory power. Consequently and following the pre-NDA meeting a discriminatory dissolution test had to be developed capable to demonstrate similarity between the extended release clinical formulation batches as well as providing an understanding of the critical quality attributes (CQA) and the critical process parameter (CPP) [3]. The ambitious time lines required the expertise and flexibility of Ardena’s resources to respond timely to the FDA requirements and finalize the NDA program as scheduled.
After reviewing the data and available samples from the immediate and extended release formulation Ardena defined a rational set of Design of Experiments. In a first step, a series of dissolution media, varying in pH and composition were performed using the USP type 2 (paddle) method at standard conditions for the immediate release as well as the newly developed extended release formulation. The dissolution screening included the quantification of the drug and its hydrolyzed derivative. The multi-media screening provided important dissolution patterns enabling the selection of two lead media compositions for the surfactant screening. After screening multiple surfactants a single surfactant was identified at two different concentrations to yield in the highest dissolution rate of the active and its degradation product. Additional comparative dissolution experiments were conducted on the available samples from the immediate and extended release formulation comparing the two media and surfactant levels which led to the selection of the most discriminatory composition. Finally, the conditions of the USP Type 2 method were modified to optimize the method by using different temperatures, vessel geometries and rotational speeds. To fulfill the regulatory requirements the method was validated according to the FDA guidelines on dissolution testing across the different product samples available. The final dissolution method than served to demonstrate the similarity between the clinical batches and provide the product as well as to serve process and product understanding for manufacturing and filing. Therefore, a risk assessment was performed and thirteen aberrant samples batches were manufactured to investigate the impact of the critical material attribute and process parameter variabilities. The comparative discriminatory dissolution data of this aberrant and the 3 registration batches are shown in figure 1. The dissolution method demonstrated the similarity of the clinical batches and identified the critical variabilities through non-similarity of the respective aberrant batch. Preferably representative aberrant batches should be produced on full scale equipment.
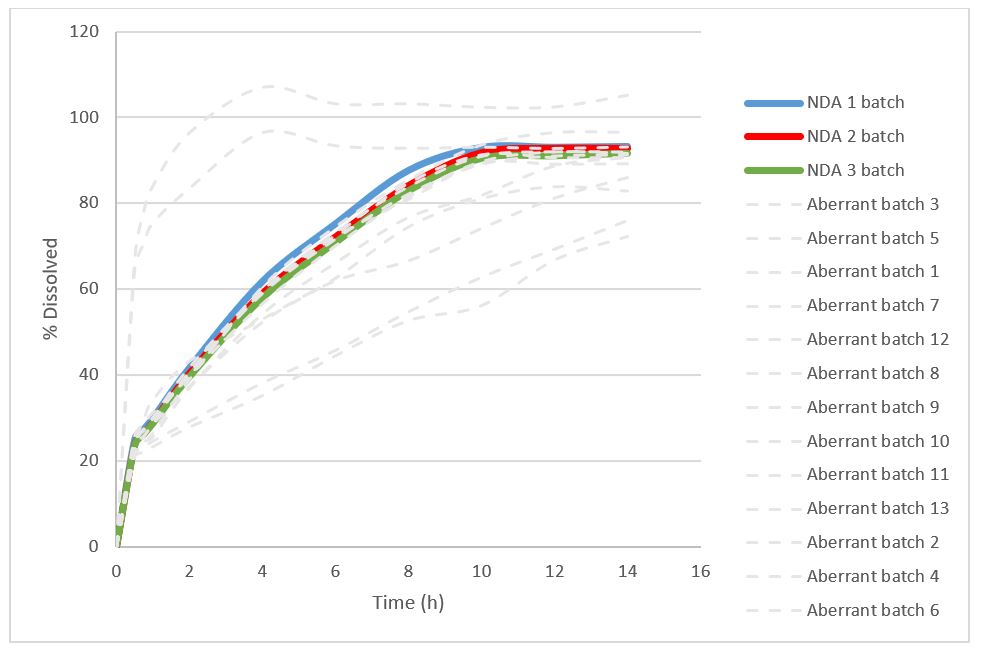
Figure 1: Dissolution profiles of aberrant core tablets and registration reference batches
The case study confirmed that advanced dissolution testing is an underestimated tool in accelerating drug product development into the clinics. The discriminatory dissolution test should be established early on in the program, ideally late phase 2 or early phase 3, to guide formulation development and clinical supplies as well as it is a tool to mitigate risk throughout the development, especially for drug repurposing or 505(b)(2) submissions [4]. Relying on Ardena’s experienced scientists provide fast and flexible resources for discriminatory dissolution test method development to support even the most ambitious development programs.
References
- Wang & Guan (2021) COVID-19 drug repurposing: A review of computational screening methods, clinical trials, and protein interaction assays. Med Res Rev. 2021;41:5–28
- Miller et al (2019) Physiologically based pharmacokinetic modelling for First-In-Human Predictions: An updated model building strategy illustrated with challenging industry case studies. Clin Pharmacokin 58:727–746
- Yu et al (2014) Understanding pharmaceutical Quality-by-Design AAPS J 16(4):771-783
- Freije et al (2020) Review of Drugs Approved via the 505(b)(2) Pathway: Uncovering drug development trends and regulatory requirements. Ther Innov 54(1):128-38
