
With increasing progress in the life sciences, we are constantly broadening our ability to treat diseases that were previously incurable. This progress however comes with new challenges due to the growing complexity of chemical and biotherapeutic products. Developing and manufacturing the compound of interest in a pure and stable form quickly and efficiently is considered to be business critical if not the decisive factor for success.
The growing complexity of chemical synthesis and biotechnological expression systems is associated with a need for more intensive fractionation and purification downstream processes to ensure sufficient product quality. Increasingly stringent quality criteria for purity are required to secure efficacy, potency, and stability, and prevent toxicity and immunogenicity, especially for biological products such as proteins, vaccines, and monoclonal antibodies. This involves the elimination of any impurities or by-products to achieve the desired compound purity while maintaining the chemical or biological stability and activity in a consistent manner.
The technical challenge of the specificity required for each product and process has led to a tremendous effort to better understand downstream processes and make them more efficient. The more technological options and processes that are available and offered, the more important the expertise in selecting, developing and scaling the most efficient approach from these options becomes.
With increasing progress in the life sciences, we are constantly broadening our ability to treat diseases that were previously incurable. This progress however comes with new challenges due to the growing complexity of chemical and biotherapeutic products. Developing and manufacturing the compound of interest in a pure and stable form quickly and efficiently is considered to be business critical if not the decisive factor for success.
The growing complexity of chemical synthesis and biotechnological expression systems is associated with a need for more intensive fractionation and purification downstream processes to ensure sufficient product quality. Increasingly stringent quality criteria for purity are required to secure efficacy, potency, and stability, and prevent toxicity and immunogenicity, especially for biological products such as proteins, vaccines, and monoclonal antibodies. This involves the elimination of any impurities or by-products to achieve the desired compound purity while maintaining the chemical or biological stability and activity in a consistent manner.
The technical challenge of the specificity required for each product and process has led to a tremendous effort to better understand downstream processes and make them more efficient. The more technological options and processes that are available and offered, the more important the expertise in selecting, developing and scaling the most efficient approach from these options becomes.
The goal is to retrieve sufficiently pure chemical and biological compounds required for the development stage, including special reagents for binding assays [read our whitepaper on bioanalysis of ADCs here] as well as GMP grade material be it an adjuvant or an active compound to support the clinical trials and commercialization. With the downstream processing contributing between 50 – 80% to the compound manufacturing costs, increasing the efficiency and simplifying the downstream process has become another important goal for drug development (Labrou, 2014).
Principles of separation and purification
The upstream processing of the chemical synthesis or biotechnology expression systems (plants, cellular, etc) is an important and critical step in the manufacture of the specific compound. The process parameters applied determine the levels and types of impurities or by-products present after the upstream step in the compound-containing substrate. The upstream process thus determines the complexity and number of fractionation and purification steps required for the downstream process.
The separation and purification of a compound is typically achieved through a variety of different physical and chemical properties which are utilized to separate the compound of interest from impurities and by-products. These compound-specific properties include size, shape, charge, isoelectric point, charge distribution, hydrophobicity, solubility, density, ligand-binding affinity, metal binding, reversible association, posttranslational modifications, and specific sequences or structures (Labrou, 2014). The major technologies used are filtration, chromatography, extraction, precipitation, and centrifugation. Filtration, extraction, precipitation, and centrifugation are mainly used for harvesting the compound-containing fraction from the up-scaling, while chromatographic and advanced filtration technologies are used for the final purification step. For complex mixtures, in particular, a scheme of different purification steps is necessary to eliminate impurities and by-products step by step to retrieve the pure compound.
Chromatography is a versatile purification technique, separating the compounds based on their size, physico-chemical, or hydrophobic/hydrophilic properties (see Table 1).
| Chromatographic purification principles | |
| Affinity (AFC) | Separation method based on a specific binding interaction between an immobilized ligand and its binding partner |
| Ion exchange (CEX, AEX) | Separation based on ions and charged molecules by affinity to the ion exchanger |
| Hydrophobic interaction (HIC) | Separation of molecules according to differences in their surface hydrophobicity |
| Size exclusion (SEC) | Separation of molecules based on their size by filtration through a gel |
| Reverse phase (RPC) | Separation by a hydrophobic stationary phase and a polar mobile phase |
| Multimodal (MMC) | Separation by more than one form of interaction between the stationary phase and analytes |
Table 1: Main chromatographic purification technologies and their purification principles
Purification by chromatography is a well understood technology, however it requires substantial expertise and knowledge as well as selective and sensitive analytical capabilities to successfully evaluate the impact of potentially critical variables (Hanke & Ottens, 2014).
Filtration methods generally consist of surface filtration, depth filtration, cross-filtration, and ultra-filtration technologies. Surface filters are single layer filters defined by their pore size. Depth filters are porous matrices purifying effectively throughout the entire depth and their efficiency can be further increased through agitation. Cross-filtration technologies such as Tangential Flow Filtration (TFF) leverage a high velocity flow tangentially across the membrane surface to purify proteins or nanoparticles. Diafiltration (DF) is a cross-flow filtration technique in which fresh solvent is added to replace the volume loss due to the filtration process. Ultra-Filtration (UF) or Nano-Filtration (NF) are pressure-driven membrane transport technologies passing mixtures through hollow fibers of membrane material. Due to the tangential filtration mode, cross-flow filtration technology prevents filter clogging or fouling which is especially important for the purification of nanoparticles (Busatto et al., 2018). This also applies to the development and commercial manufacturing of new vaccine and vaccine adjuvant nanoparticle products (Pires et al., 2023).
Approaches in the development of fractionation and purification towards downstream processing
A systematic yet practical approach for fractionation and purification is required when starting the process development to achieve the desired objectives and milestones in each phase. Table 2 shows some important objectives of fractionation and purification at each stage and demonstrates that fractionation and purification should start at the drug discovery phase and continue through to a validated and commercially viable downstream process.
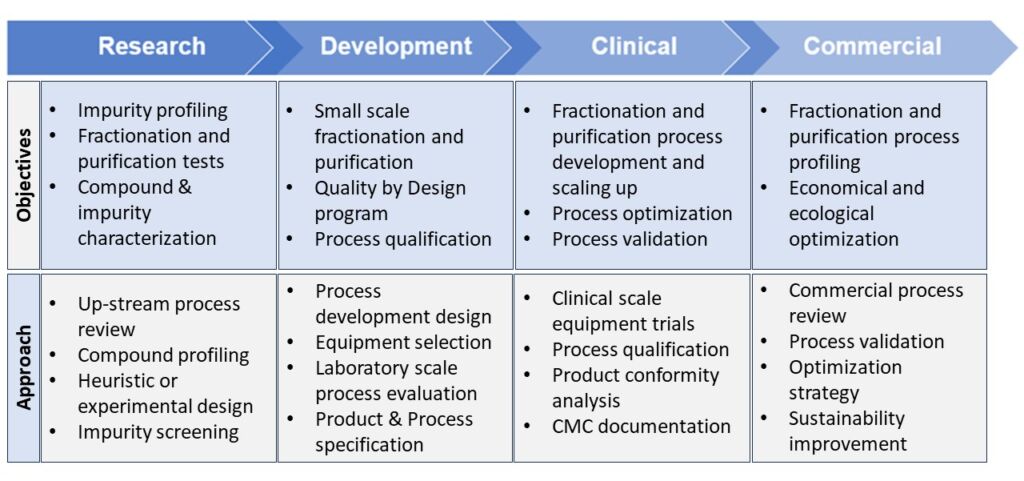
Table 2: Objectives and approaches for fractionation and purification throughout the drug discovery and development phases and seamless transition into the commercial downstream process
For each compound, be it derived from chemical synthesis, biotechnological expression system, or natural origin, a rational and product specific development strategy is required. After reviewing the synthesis or expression and product characteristics a universal or process specific heuristic approach can be used effectively for known products and processes (Pfister et al., 2017). In case of new products and processes, an experimental approach may be more target-oriented and can be performed in a high throughput manner. These high throughput process development (HTPD) tools consist of parallel, miniaturized automated systems which can be used for the upstreaming and downstreaming of the product development (Baumann & Hubbuch, 2017) .
Standard or platform approaches for purification have also been suggested for monoclonal antibodies, which require only limited development work and have a high probability to achieve the degree of purity required (Shukla et al., 2007). While they might serve the purpose during the development stage, they might not provide the most efficient option for the commercial scale due to the expensive Protein A chromatography step. To overcome this issue techniques like aqueous two-phase separation technique are under investigation (Kruse et al., 2020).
The need to increase efficiency in separation and purification
In the early phases of a development program, especially when entering into a phase 1 clinical trial, downstream processing is time-critical and the speed of its development is a top priority. However, in the later development phase the success-critical factors of the efficiency and scalability of the process come to the fore.
A heuristic approach coupled with HTPD technology based on small scale automatic dispensing and sampling equipment have proven to be extremely powerful in accelerating downstream development in the early phase. For example, preparative high-performance liquid chromatography (HPLC) has evolved as a scalable purification and polishing technology throughout the product life cycle (De Luca et al., 2021). HTPD technology can also be used to generate large sets of experimental data to support development according to the quality-by-design (QbD) principles or a statistical and predictive model-based approach (Keulen et al., 2022). In order to overcome some of the limitations of the purification of peptides by chromatography catch and release (C&R) methods based on a specific combination of base-labile cleavable linkers with oxime-based and hydrazone-based ligation chemistry are under evaluation but these require more work to further reduce off-target reactions (Reimann et al., 2019).
The growing public awareness of climatic change and environmental protection has put pressure on the pharmaceutical industry, the manufacturing processes and products. Sustainability is therefore not only a question of corporate identity but also of a company’s profitability. Sustainability in biopharmaceutical manufacturing must be considered across the entire process, with the downstream processing playing a special role. Improvements can be achieved by using less critical solvents, whereas optimization of processes and procedures can achieve solvent savings of up to 90% (Ferrazzano et al., 2022). These savings are achievable because batch-based processes are typically affected by a yield-purity trade-off, due to overlapping regions where either some the by-products will be carried over or product will be lost due to the narrow fractionation or purification window (De Luca et al., 2020; Ferrazzano et al., 2022). With the technological advances in downstream processing and the use of QbD principles, predictive models can be used to establish a PAT-based close-loop control (Gerzon et al., 2022) which increasingly enables the conversion of the upstream and the downstream processes into a continuous process (Behere & Yoon, 2020). It is estimated that continuous bioprocessing could save approximately 55% of the costs compared to traditional batch manufacturing (Walther et al., 2015). Continuous processing is supported and promoted by the FDA through the introduction of guidelines to improve manufacturing and patient access to innovative biotherapeutics (Fisher et al., 2019).
While the development of a continuous manufacturing process for clinical phases 1 and 2 is disproportionate, individual downstream process steps can already be carried out continuously or later transferred into continuous processing mode. Cross-flow filtration technologies such as TFF, DF, UF and NF are process technologies serving small and large scale as well as batch and continuous processing options (David et al., 2020). Besides their efficient purification these technologies are also suited to producing higher concentrations of biochemical compounds as well as performing the essential buffer changes at the end of the purification step (De Luca et al., 2020).
Enhancing compound concentration and stability can be achieved by eliminating reactive or catalytic impurities through purification, which can be further optimized through an additional lyophilization process. This may be especially important for overcoming the transport and storage issues associated with vaccines for tropical diseases or third world use e.g. Covid vaccines (Crommelin et al., 2021).
Case studies
Ardena has carried out several upstream and downstream programs in partnership with small and large enterprises to support chemical and biological development programs from the very early phases through to commercial scale.
In a phase-appropriate development approach, especially in early phases when the demand for drug substance is still limited, the common drawbacks of chromatographic methods such as cost and throughput may be offset by benefits: time of delivery and purity. In critical cases where the impurity profile is challenging and development of the chemical process is time consuming, the application of chromatographic methods provides a clear advantage.
In one of our projects a key starting material for GMP manufacturing was prepared via a photo-isomerization reaction on a steroid substrate which produced a mixture of the target compound together with a range of impurities. The mixture contained about 50% of the desired material hence crystallization was not feasible. Gravity chromatography was developed and scaled which allowed the production of the material on a multikilogram scale.

With the successful launch of Covid vaccines mRNA delivery technology has become one of the most exciting and promising approaches for innovative therapies. For the stabilization and transfection of the mRNA, specifically designed ionic lipids or lipoids are an essential part of the delivery system (Zhang et al., 2021). Using an innovative cationic lipidoid derivative identified by a company in a screening program, a complex ionic lipoid was custom-synthesized and more than 500 g of GMP grade material was produced. Upscaling required the development of an upstream synthesis route and a downstream purification process that would meet the efficiency requirements of the commercial manufacturing process. The development of the synthesis was oriented towards high selectivity and avoidance of impurities that are difficult to separate. Through a purification step of an intermediate in the synthesis by using reverse phase chromatography, the upstream product was obtained with high purity. For the final downstream processing, flash chromatography was successfully applied together with preparative HPLC coupled to UV-Vis and Evaporative Light Scattering detectors. The equipment used to process 500 g of GMP grade of the ionic lipoid, a Buchi C850 system, facilitated both rapid production of sufficient quantities for subsequent clinical development and seamless transfer to larger-scale equipment for commercial volumes.
Fractionation and purification of sensitive nanoparticles (e.g. vaccines, liposomes etc.) is a growing field of advanced drug delivery. One example is a new vaccine adjuvant which was developed by one of our customers, targeting the immune stimulatory complexes (ISCOMs) to increase the immune response of protein-based vaccines. This matrix is based on saponins of natural origin. An extract is fractionated and the fractions are collected and lyophilized. The fractions are formulated with phospholipids and cholesterol to retrieve two distinct nanoparticles (Bengtsson et al., 2011; Magnusson et al., 2018).
During a fast-track development project by a major healthcare company, purification of an innovative Manganese-based contrast agent became the bottleneck in development and threatened to significantly delay entry into clinical development. Following the upstream process, the compound containing aqueous solution was passed through a nanofiltration TFF process to separate molecules with a molecular weight of 100-250 Da from the compound. Ion-exchange chromatography was selected due to the affinity profile of the compound and a stationary phase, mobile phase, and gradient were developed to retrieve high yields of pure GMP grade compound from the pre-purified solution. Due to the instability of the compound in solution, a lyophilization step was added to provide the GMP grade compound in a stable solid form. The downstream process developed leveraged prior experience on a volume-efficient and scalable approach toward commercial manufacturing.
Conclusion
Purification plays an important role at multiple stages of drug development and manufacturing of complex drug synthesis and biotechnology expression systems. Developing an effective and efficient downstream purification process is upstream and product dependent and requires individually tailored purification schemes.
It is therefore important to choose the right approach for the upstream and downstream processes that ensures safe, reproducible purification and can be efficiently transferred to a manufacturing scale. It is important to use established processes whenever possible and to avoid complex and lengthy purification procedures and overengineering.
The challenge of considering commercial manufacturing early in the development phase is often underestimated. The difficulty arises from the wide variety of parameters and equipment variables which are being tested during development without consideration for the readiness of the future commercial manufacturing facility (Chahar et al., 2020; Matte, 2022).
Science and knowledge-based approaches coupled with strong analytical capabilities accelerate the time critical downstream process development including the lay out towards the most efficient commercial scale purification. Depending on their internal infrastructure and organizational setup, companies can greatly benefit from outsourcing development and manufacturing processes.
References
Baumann, P., & Hubbuch, J. (2017). Downstream process development strategies for effective bioprocesses: Trends, progress, and combinatorial approaches. In Engineering in Life Sciences (Vol. 17, Issue 11, pp. 1142–1158). Wiley-VCH Verlag. https://doi.org/10.1002/elsc.201600033
Behere, K., & Yoon, S. (2020). Chromatography bioseparation technologies and in-silico modelings for continuous production of biotherapeutics. Journal of Chromatography A, 1627. https://doi.org/10.1016/j.chroma.2020.461376
Busatto, S., Vilanilam, G., Ticer, T., Lin, W. L., Dickson, D. W., Shapiro, S., Bergese, P., & Wolfram, J. (2018). Tangential flow filtration for highly efficient concentration of extracellular vesicles from large volumes of fluid. Cells, 7(12). https://doi.org/10.3390/cells7120273
Chahar, D. S., Ravindran, S., & Pisal, S. S. (2020). Monoclonal antibody purification and its progression to commercial scale. In Biologicals (Vol. 63, pp. 1–13). Academic Press. https://doi.org/10.1016/j.biologicals.2019.09.007
Crommelin, D. J. A., Anchordoquy, T. J., Volkin, D. B., Jiskoot, W., & Mastrobattista, E. (2021). Addressing the Cold Reality of mRNA Vaccine Stability. Journal of Pharmaceutical Sciences, 110(3), 997–1001. https://doi.org/10.1016/j.xphs.2020.12.006
David, L., Schwan, P., Lobedann, M., Borchert, S. O., Budde, B., Temming, M., Kuerschner, M., Alberti Aguilo, F. M., Baumarth, K., Thüte, T., Maiser, B., Blank, A., Kistler, V., Weber, N., Brandt, H., Poggel, M., Kaiser, K., Geisen, K., Oehme, F., & Schembecker, G. (2020). Side-by-side comparability of batch and continuous downstream for the production of monoclonal antibodies. Biotechnology and Bioengineering, 117(4), 1024–1036. https://doi.org/10.1002/bit.27267
De Luca, C., Felletti, S., Lievore, G., Chenet, T., Morbidelli, M., Sponchioni, M., Cavazzini, A., & Catani, M. (2020). Modern trends in downstream processing of biotherapeutics through continuous chromatography: The potential of Multicolumn Countercurrent Solvent Gradient Purification. In TrAC – Trends in Analytical Chemistry (Vol. 132). Elsevier B.V. https://doi.org/10.1016/j.trac.2020.116051
De Luca, C., Lievore, G., Bozza, D., Buratti, A., Cavazzini, A., Ricci, A., Macis, M., Cabri, W., Felletti, S., & Catani, M. (2021). Downstream processing of therapeutic peptides by means of preparative liquid chromatography. In Molecules (Vol. 26, Issue 15). MDPI AG. https://doi.org/10.3390/molecules26154688
Ferrazzano, L., Catani, M., Cavazzini, A., Martelli, G., Corbisiero, D., Cantelmi, P., Fantoni, T., Mattellone, A., De Luca, C., Felletti, S., Cabri, W., & Tolomelli, A. (2022). Sustainability in peptide chemistry: Current synthesis and purification technologies and future challenges. In Green Chemistry (Vol. 24, Issue 3, pp. 975–1020). Royal Society of Chemistry. https://doi.org/10.1039/d1gc04387k
Fisher, A. C., Kamga, M. H., Agarabi, C., Brorson, K., Lee, S. L., & Yoon, S. (2019). The Current Scientific and Regulatory Landscape in Advancing Integrated Continuous Biopharmaceutical Manufacturing. In Trends in Biotechnology (Vol. 37, Issue 3, pp. 253–267). Elsevier Ltd. https://doi.org/10.1016/j.tibtech.2018.08.008
Gerzon, G., Sheng, Y., & Kirkitadze, M. (2022). Process Analytical Technologies – Advances in bioprocess integration and future perspectives. Journal of Pharmaceutical and Biomedical Analysis, 207. https://doi.org/10.1016/j.jpba.2021.114379
Hanke, A. T., & Ottens, M. (2014). Purifying biopharmaceuticals: Knowledge-based chromatographic process development. In Trends in Biotechnology (Vol. 32, Issue 4, pp. 210–220). Elsevier Ltd. https://doi.org/10.1016/j.tibtech.2014.02.001
Keulen, D., Geldhof, G., Bussy, O. Le, Pabst, M., & Ottens, M. (2022). Recent advances to accelerate purification process development: A review with a focus on vaccines. Journal of Chromatography A, 1676. https://doi.org/10.1016/j.chroma.2022.463195
Kruse, T., Kampmann, M., Rüddel, I., & Greller, G. (2020). An alternative downstream process based on aqueous two-phase extraction for the purification of monoclonal antibodies. Biochemical Engineering Journal, 161. https://doi.org/10.1016/j.bej.2020.107703
Labrou, N. E. (2014). Protein Purification: An overview. In N. E. Labrou (Ed.), Methods in Molecular Biology: Protein Downstream Processing (Vol. 1129, pp. 3–10). http://www.springer.com/series/7651
Lövgren Bengtsson, K., Morein, B., & Osterhaus, A. D. (2011). ISCOM technology-based Matrix MTM adjuvant: Success in future vaccines relies on formulation. In Expert Review of Vaccines (Vol. 10, Issue 4, pp. 401–403). https://doi.org/10.1586/erv.11.25
Magnusson, S. E., Altenburg, A. F., Bengtsson, K. L., Bosman, F., de Vries, R. D., Rimmelzwaan, G. F., & Stertman, L. (2018). Matrix-MTM adjuvant enhances immunogenicity of both protein- and modified vaccinia virus Ankara-based influenza vaccines in mice. Immunologic Research, 66(2), 224–233. https://doi.org/10.1007/s12026-018-8991-x
Matte, A. (2022). Recent Advances and Future Directions in Downstream Processing of Therapeutic Antibodies. In International Journal of Molecular Sciences (Vol. 23, Issue 15). MDPI. https://doi.org/10.3390/ijms23158663
Pfister, D., David, L., Holzer, M., & Nicoud, R. M. (2017). Designing affinity chromatographic processes for the capture of antibodies. Part I: A simplified approach. Journal of Chromatography A, 1494, 27–39. https://doi.org/10.1016/j.chroma.2017.02.070
Pires, I. S., Ni, K., Melo, M. B., Li, N., Ben-Akiva, E., Maiorino, L., Dye, J., Rodrigues, K. A., Yun, D. S., Kim, B., Hosn, R. R., Hammond, P. T., & Irvine, D. J. (2023). Controlled lipid self-assembly for scalable manufacturing of next-generation immune stimulating complexes. Chemical Engineering Journal, 464. https://doi.org/10.1016/j.cej.2023.142664
Reimann, O., Seitz, O., Sarma, D., & Zitterbart, R. (2019). A traceless catch-and-release method for rapid peptide purification. Journal of Peptide Science, 25(1). https://doi.org/10.1002/psc.3136
Shukla, A. A., Hubbard, B., Tressel, T., Guhan, S., & Low, D. (2007). Downstream processing of monoclonal antibodies-Application of platform approaches. In Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences (Vol. 848, Issue 1, pp. 28–39). https://doi.org/10.1016/j.jchromb.2006.09.026
Walther, J., Godawat, R., Hwang, C., Abe, Y., Sinclair, A., & Konstantinov, K. (2015). The business impact of an integrated continuous biomanufacturing platform for recombinant protein production. Journal of Biotechnology, 213, 3–12. https://doi.org/10.1016/j.jbiotec.2015.05.010
Zhang, Y., Sun, C., Wang, C., Jankovic, K. E., & Dong, Y. (2021). Lipids and Lipid Derivatives for RNA Delivery. In Chemical Reviews (Vol. 121, Issue 20, pp. 12181–12277). American Chemical Society. https://doi.org/10.1021/acs.chemrev.1c00244
