The chemical synthesis and supply of a drug substance is a time critical step in the process of defining and selecting a lead drug candidate for progress into a toxicology program and subsequently into first-in-human clinical trials. The presence of impurities can hinder toxicology results and delay entry into the clinic therefore it is important to identify and mitigate this risk. There are multiple potential causes of the formation or introduction of impurities regardless of the synthesis route used, including the materials used, the process conditions, potential cross-contamination, etc., and these can lead to several types of impurities.
The recent recalls of sartans and other medicinal products due to the presence of N‑nitrosamine impurities, increased the awareness of the impact of material selection, process changes, and cross-contamination, as well as the limitations of the existing analytical methods used. Many N‑nitrosamines are known to be carcinogenic to animals and are reasonably anticipated to be human carcinogens.
Authors: Lieven Van Vooren – Scientific Director, Arno Vermote – Senior CMC Writer
The chemical synthesis and supply of a drug substance is a time critical step in the process of defining and selecting a lead drug candidate for progress into a toxicology program and subsequently into first-in-human clinical trials. The presence of impurities can hinder toxicology results and delay entry into the clinic therefore it is important to identify and mitigate this risk. There are multiple potential causes of the formation or introduction of impurities regardless of the synthesis route used, including the materials used, the process conditions, potential cross-contamination, etc., and these can lead to several types of impurities (see table 1).

Table 1: Types of impurities in drug substance
The case of N-nitrosamine impurities
The recent recalls of sartans and other medicinal products due to the presence of N‑nitrosamine impurities, increased the awareness of the impact of material selection, process changes, and cross-contamination, as well as the limitations of the existing analytical methods used. Many N‑nitrosamines are known to be carcinogenic to animals and are reasonably anticipated to be human carcinogens.
N‑nitrosamines can be formed by a nitrosating reaction between an amine (secondary, tertiary, or quaternary amines) and a nitrosating agent (e.g., nitrite salts) under acidic reaction conditions. Nitrosating reactions can potentially occur during production under certain conditions and when nitrogen-containing solvents, reagents, and other raw materials are used. In addition impurities can be carried over during the manufacturing process when using already contaminated equipment or reagents. In cases where N-nitrosamines can form or are carried over during production, the impurities should be controlled and removed during the manufacturing process.
Regulatory requirements and documentation
Since this discovery of N‑nitrosamines in pharmaceuticals, regulatory authorities in Europe and the U.S. have undertaken numerous activities to assess the extent, understand the root cause, establish expert working groups, and develop appropriate N‑nitrosamine impurity guidelines.
One of the essential parts of Health Authority regulations is a risk assessment of the chemical synthesis. Based on a solid understanding of the root causes of formation of N‑nitrosamines and relevant experience in chemical synthesis, the occurrence of N‑nitrosamines in the drug substance can be avoided and/or controlled sufficiently. Ardena ensures compliance with these guidelines through coordinated action among its Centers of Excellence. The Ardena Dossier Development team has a critical role to play in all this and has written over 100 nitrosamine risk assessment reports for multiple customers.
These risk assessments consist of 3 steps.
The first step is to perform a comprehensive risk evaluation to identify if there is a risk of N-nitrosamines being present in the API and/or finished product. As illustrated in Figure 1, both the potential formation of N‑nitrosamines and the cross-contamination risk are scrutinized.
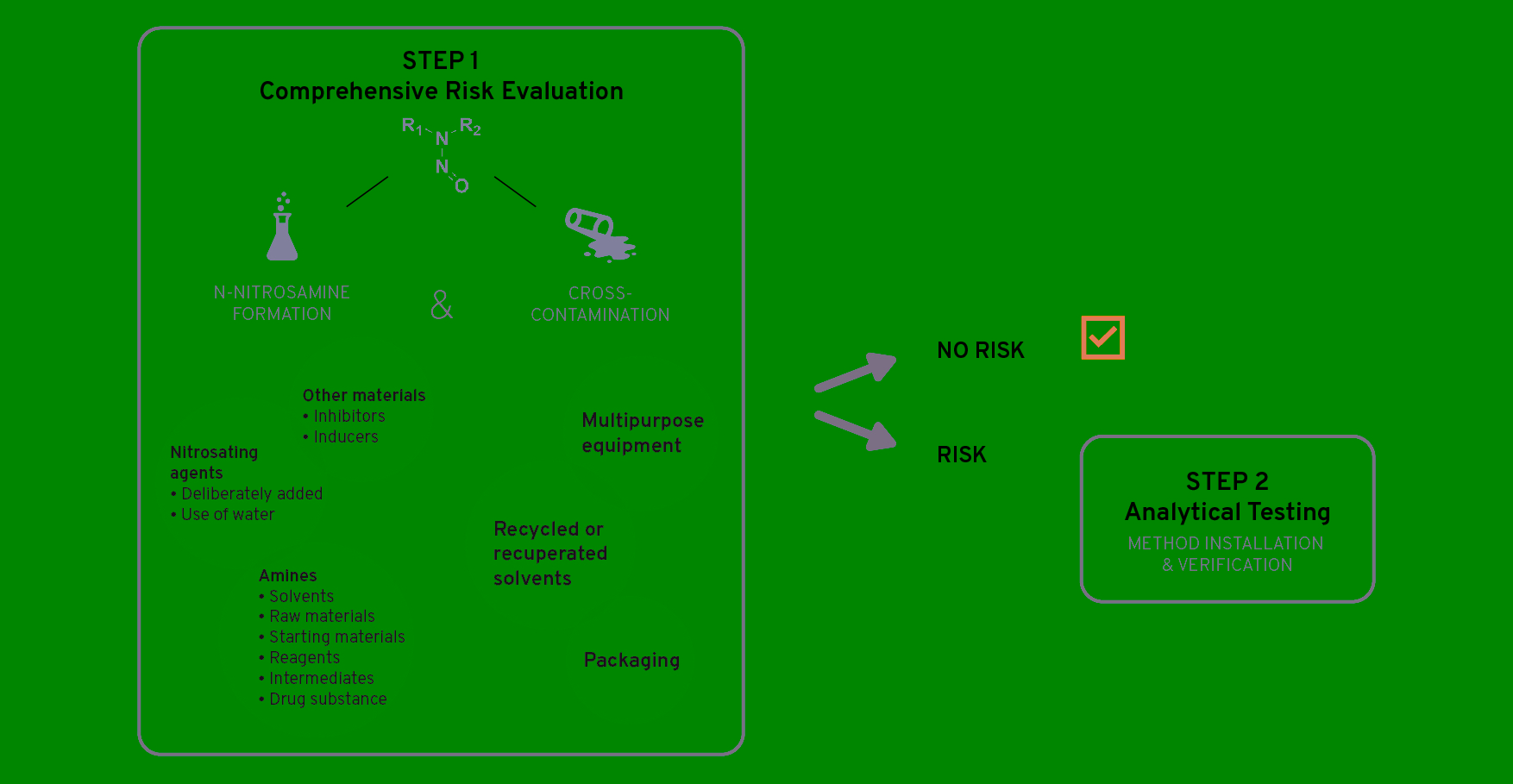
Figure 1: N-nitrosamine investigation – Step-by-step plan, Step 1
The presence of amines and nitrosating agents in combination with the reaction conditions and potential inducers or inhibitors of nitrosamine formation are examined. Potential cross-contamination is evaluated by looking at recycled materials, multipurpose equipment, packaging, etc.
As illustrated in figure 2, if a risk is identified, confirmatory analytical testing (i.e., Step 2) is required in order to confirm or refute the presence of nitrosamines. If the presence of nitrosamines is confirmed, effective risk mitigating measures should be implemented (i.e., Step 3).
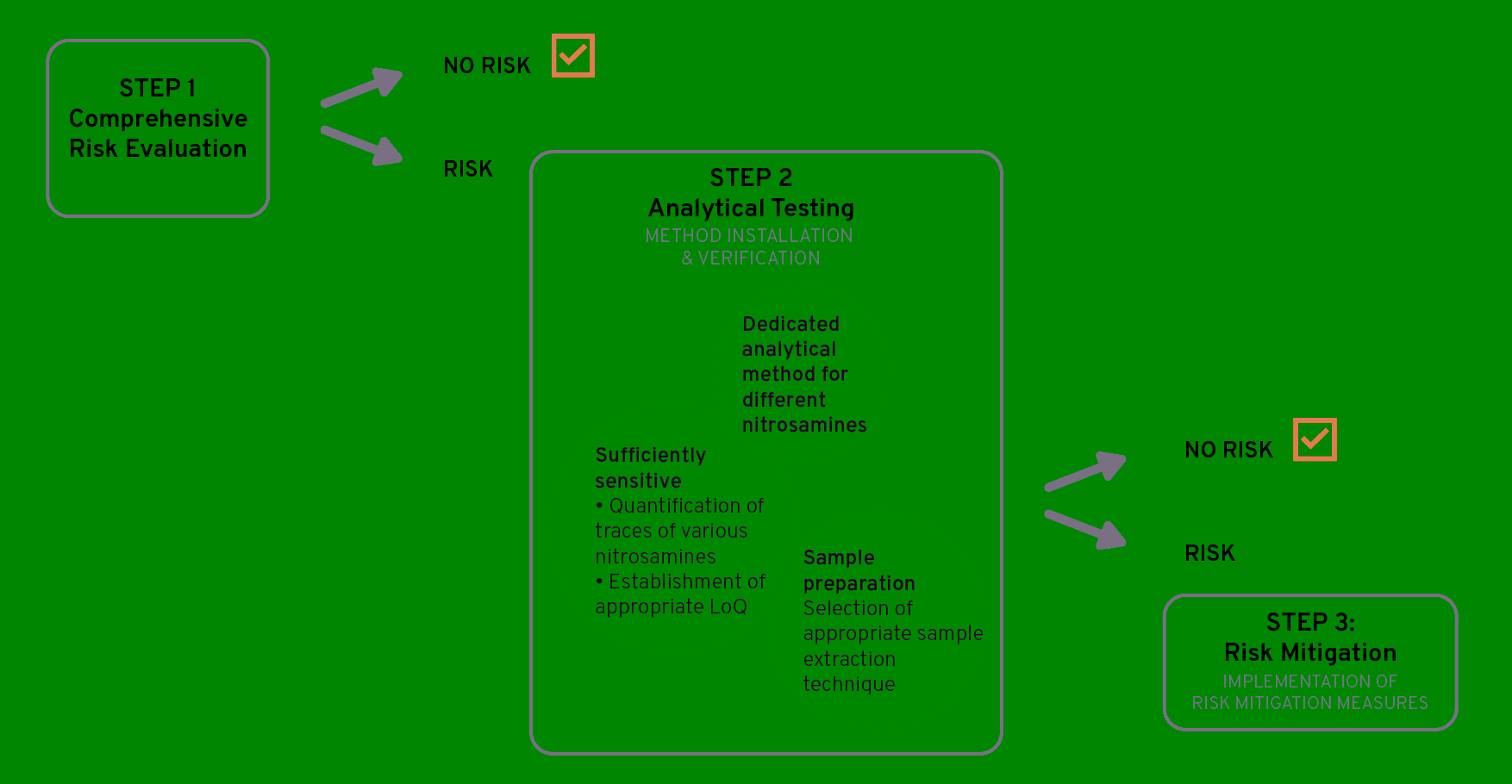
Figure 2: N-nitrosamine investigation – Step-by-step plan, Step 2 & 3
Proactive risk mitigation – Case study
N-nitrosamine risk assessments are necessary not only for commercial products but are also highly recommended during the development phase to investigate potential contamination.
The importance of assessing potential N‑nitrosamine contamination early in development can be illustrated by a typical customer request that Ardena addressed successfully In a drug substance synthesis project, one of the raw materials was a piperidine derivative. The manufacturer of this raw material indicated that during the synthesis piperidine-like structure secondary and/or tertiary amines were used in the same and subsequent steps as nitrosating agents. As the raw material was introduced in one of the final steps of the drug substance synthesis, it could not be excluded that N‑nitrosamines would be present in the eventual drug substance. In addition, theoretical purging calculations did not conclude that the potential nitrosamines were sufficiently purged. As a proactive mitigation plan in order to avoid analytical testing (i.e., Step 2 in Figure 2), the synthesis route was modified with the introduction of the piperidine derivative at the start of the synthesis as well as the addition of an extra crystallization step in the end.
By monitoring this potential nitrosamine formation early in development, we were able to react quickly and immediately choose the most suitable synthesis route for further development, without the risk of having to change the synthesis later in development.
To ensure the rapid initiation of toxicology studies and entry into the clinical phase, it is not sufficient to rely only on controlling the impurities below phase-appropriate specification limits. The synthesis of the drug substance must also be well understood, controlled, and accompanied by phase-appropriate analytical methods. To meet the regulatory requirements comprehensive documentation is essential as well as confidence in the scalability of the chemical synthesis to support the later clinical phases and commercialization. More information about this in our whitepaper titled ‘Phase-appropriate analytical method development’. To avoid undesirable and unexpected impurities during later phases, these should be considered during synthesis, analytical method development, and regulatory documentation.
The relevant CMC documentation and if necessary direct support in FDA, pre-IND, or EMA scientific advice meetings is provided by Ardena’s regulatory experts. More information about this in our whitepaper titled ‘Leverage the power of CMC in early drug development’.
N-nitrosamines – Acceptable limits in medicinal products
A specific challenge of N‑nitrosamine impurities is their very low acceptable levels, as defined by their acceptable daily intake (ADI; Table 2). Consequently they depend on the targeted daily clinical dose which can quickly exceed the ADI.
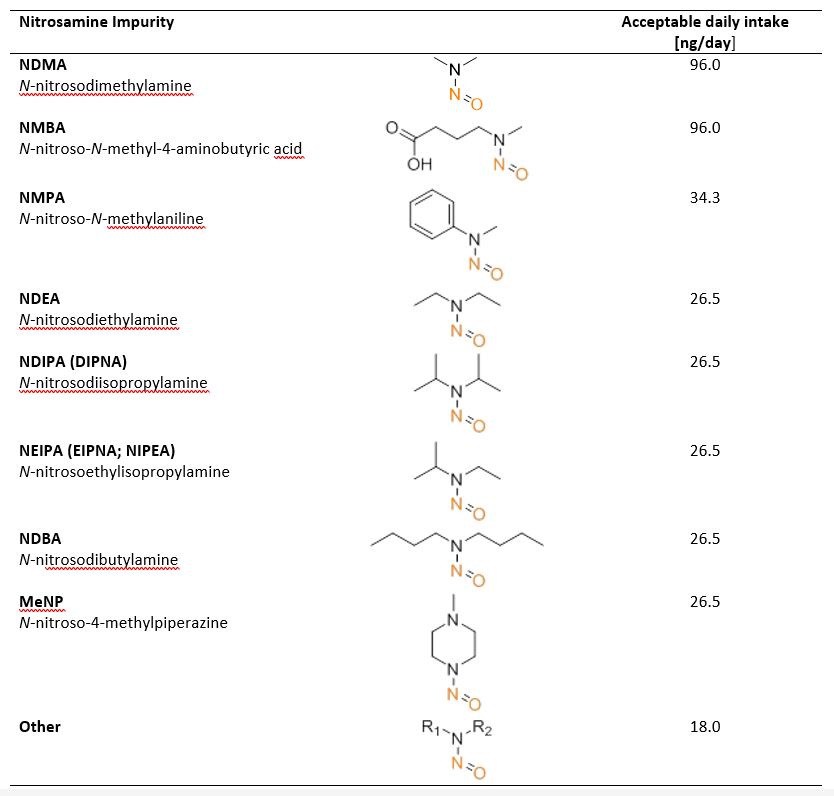
Table 2: Some specific N‑nitrosamines and their acceptable daily intake (ADI)
If multiple different N‑nitrosamines are present, the ADI limit must be calculated on the sum of all N‑nitrosamines and their specific ADIs. Such low thresholds imply that even traces from chemical precursors or non-optimized process conditions can lead to limits being exceeded. Since the FDA guidelines require analytical methods for N‑nitrosamines which have limits of quantitation (LOQ) in the parts-per-billion (ppb) range to ensure that the ADI is not exceeded, the management of N‑nitrosamine impurities must be part of the risk assessment and mitigation process.
An integrated approach to manage N‑nitrosamine impurities in clinical drug supplies
As illustrated above, by carrying out a risk assessment before deciding on the synthesis route of a new drug substance Ardena ensures that impurities are reduced to a minimum and that a targeted analysis is carried out regarding the possible impurities. This risk assessment is later formalized to support the regulatory documentation according to the ICH Q11 guideline.
By their nature N‑nitrosamines are impurities that occur in the ppm range and can be present in various analytical artifacts. In addition to the regulatory need to determine the presence of N-nitrosamines in marketed pharmaceutical products, there is a demand for appropriately sensitive and specific analytical methods. The challenge is that the methods must be developed and qualified specifically for each drug substance and/or drug product. Analytical method development includes appropriate sample preparation, separation, and detection of individual N‑nitrosamines. In the case of N‑nitrosamines, thermal and pH stability must be taken into account during sample preparation. Due to the extremely low concentration of the N‑nitrosamine impurities extraction methods are also usually used, mostly solid phase extraction (SPE), liquid-liquid extraction (LLC), or direct liquid extraction (DLE). The resulting sample is then further separated by gas chromatography (GC) or liquid chromatography (LC) and the individual N‑nitrosamine artifacts determined.
The limit of quantification (LoQ) plays an important role as it provides the minimum level at which an analyte can be quantified with acceptable accuracy, and it should be used for N-nitrosamine testing and decision-making
To cover the full range of possible N‑nitrosamines, Ardena develops a product-specific approach from one or several analytical methods that provide the highest possible resolution while being cost- and time-efficient.
Conclusion
Ardena’s integrated science-driven and concerted approach in drug substance synthesis, analytical method development, and regulatory documentation assures compliance with the latest N‑nitrosamine impurity guidance and requirements.
References
- Alsante et al (2014). Recent trends in product development and regulatory issues on impurities in active pharmaceutical ingredient (API) and drug products. Part 1: Predicting degradation related impurities and impurity considerations for pharmaceutical dosage forms. AAPS PharmSciTech 15(1): 198-212.
- Bharate SS (2021). Critical analysis of drug product recalls due to nitrosamine
impurities. J Med Chem 64(6): 2923-2936. - Brown et al (2016). Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone? J Med Chem 59(10): 4443-4458.
- Charoo et al (2019). Lesson learnt from recall of valsartan and other angiotensin II receptor blocker drugs containing NDMA and NDEA impurities. AAPS PharmSciTech 20(5): 166.
- Chidella KS et al (2021). Ultra-sensitive LC-MS/MS method for the trace level quantification of six potential genotoxic nitrosamine impurities in telmisartan. Am J Anal Chem 12: 227-240.
- EMA (2020). Assessment Report. Nitrosamine impurities in human medicinal products. EMA/369136/2020. https://www.ema.europa.eu/en/documents/referral/nitrosamines-emea-h-a53-1490-assessment-report_en.pdf
- EMA (2020). Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 12-15 October 2020 https://www.ema.europa.eu/en/news/meeting-highlights-committee-medicinal-products-human-use-chmp-12-15-october-2020
- EMA (2021). European Medicines Regulatory Network approach for the implementation of the CHMP Opinion pursuant to Article 5(3) of Regulation (EC) No 726/2004 for nitrosamine impurities in human medicines. EMA/425645/2020. https://www.ema.europa.eu/en/documents/referral/european-medicines-regulatory-network-approach-implementation-chmp-opinion-pursuant-article-53/2004-nitrosamine-impurities-human-medicines_en.pdf
- EMA (2022). Fifth Nitrosamine Implementation Oversight Group (NIOG) meeting. https://www.ema.europa.eu/en/events/fifth-nitrosamine-implementation-oversight-group-niog-meeting
- EMA (2022). Questions and answers for marketing authorisation holders/applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 referral on nitrosamine impurities in human medicinal products. EMA/409815/2020. https://www.ema.europa.eu/en/documents/referral/nitrosamines-emea-h-a53-1490-questions-answers-marketing-authorisation-holders/applicants-chmp-opinion-article-53-regulation-ec-no-726/2004-referral-nitrosamine-impurities-human-medicinal-products_en.pdf
- FDA (2019). GC/MS headspace method for detection of NDMA in valsartan drug substance and drug products. https://www.fda.gov/media/115965/download
- FDA (2021). Control of nitrosamine impurities in human drugs. Guidance for industry. February 2021. https://www.fda.gov/media/141720/download
- FDA (2021). Updates on possible mitigation strategies to reduce the risk of nitrosamine drug substance-related impurities in drug products. https://www.fda.gov/drugs/drug-safety-and-availability/updates-possible-mitigation-strategies-reduce-risk-nitrosamine-drug-substance-related-impurities
- Fritzsche M et al (2022). NDMA analytics in metformin products: Comparison of methods and pitfalls. Eur J Pharm Sci 168: 106026.
- ICH Q11 (2012). Development and manufacture of drug substances (chemical entities and biotechnological/biological entities): https://database.ich.org/sites/default/files/Q11%20Guideline.pdf
- King et al (2020). Ranitidine – investigations into the root cause for the presence of N‑nitroso‑N,N‑dimethylamine in ranitidine hydrochloride drug substances and associated drug products. Org Process Res Dev 24(12): 2915−2926.
- Leistner et al (2020). Risk assessment report of potential impurities in cetirizine dihydrochl J Pharm Biomed Anal 189: 113425.
- Parr MK & Joseph JF (2019). NDMA impurity in valsartan and other pharmaceutical products:
Analytical methods for the determination of N-nitrosamines. J Pharm Biomed Anal 164: 536–549.