Advancing Pharmaceutical Production: The Benefits of Flow Manufacturing for Nanomedicines
Nanomedicines demand tight control over particle properties, reproducibility, and scalability, yet conventional batch methods often struggle to deliver all three.
In this whitepaper, Ardena’s nanomedicines experts Mark van Eldijk and Andy Bänziger explore how flow manufacturing can enable more consistent nanoparticle production, simplify scale-up, and support robust development pathways from early-stage research through GMP manufacturing.
The paper includes a practical case study using a multi-inlet vortex mixer (MIVM) for liposome production, showing how key process parameters influence particle size and polydispersity.
Download the whitepaper to learn:
Why flow manufacturing improves reproducibility compared to batch processes
How continuous processing supports scalable nanoparticle production
A real formulation case study demonstrating parameter-driven control
Get the whitepaper by filling in the form.
Thank you for your interest in our scientific content.
Evolution of Stability Guidelines: From the ICH Q1A-F Series to the Consolidated ICH Q1 Guideline
The ICH has released a draft consolidated guideline (ICH Q1) that unifies the previous Q1A–Q1F series into a single global standard. This revision simplifies stability requirements, integrates lifecycle management, and expands the scope to advanced therapies and drug-device combinations.
This whitepaper provides a clear overview of the key changes — from predictive tools and modeling to stability expectations for ATMPs and critical auxiliary materials.
Download the full whitepaper to explore how the new guideline impacts your development and regulatory strategy
Thank you for your interest in our scientific content.
Deciphering the Complex Characteristics of Nanomedicine
Nanomedicines are redefining pharmaceutical innovation, but their complexity demands advanced analytical solutions. In this whitepaper, Ardena explores the analytical and regulatory challenges of characterizing nanoparticle-based drug delivery systems, and how state-of-the-art techniques like AF4-MALS-DLS can provide critical insights into particle size, morphology, stability, and drug loading.
Gain a deeper understanding of:
– How to identify and validate Critical Quality Attributes (CQAs)
– The limitations of conventional techniques like DLS
– Regulatory trends and the path toward standardization
Read the full whitepaper.
Thank you for your interest in our scientific content.
Late-Stage Development and Manufacturing of Novel Excipients for Precision Nanomedicines and Targeted Therapies
Excipients are no longer passive ingredients—they’re critical to the success of precision drug delivery systems. In this whitepaper, Ardena shares insights into the development and GMP manufacturing of two novel phospholipids used in nanomedicine applications, including oncology and thermosensitive liposomes.
Gain a deeper understanding of:
– Control strategies for novel excipients
– Overcoming analytical and scalability challenges
– The regulatory implications of excipient innovation
Read the full whitepaper.
Thank you for your interest in our scientific content.
Innovations in API Manufacturing of Small Molecule Drugs
How can the pharmaceutical industry improve efficiency, sustainability, and adaptability in API manufacturing?
In this whitepaper, Dirk-Jan van Zoelen, PhD, explores how applying the 6M framework—Manpower, Method, Machine, Material, Measurement, and Mother Nature—enables scientific and operational innovation across the development and scale-up of small molecule APIs.
Learn how emerging technologies like AI, flow chemistry, enzymatic catalysis, and greener synthesis approaches are helping reduce costs and environmental impact—while ensuring quality and regulatory robustness.
Fill in the form to access the full whitepaper.
Thank you for your interest in our scientific content.
(Cross-)Contamination Control through Effective Equipment Cleaning
Drug discovery and the scale-up of newly developed manufacturing processes represent an exciting and dynamic field for academic scientists, startups, and small to large pharmaceutical companies. The scale-up to clinical batch sizes, in particular, poses challenges as lab-scale manufacturing expands to batch sizes in the multi-kg range, requiring further optimization of manufacturing processes. Due to the limited and often unknown safety profiles of new compounds, this scale-up is conducted in specialized facilities equipped with adequate contingency measures.
To ensure patient safety, a key issue in the manufacture of active pharmaceutical ingredients (APIs) in multi-purpose, multi-product manufacturing equipment is the adequate management of potential (cross-)contamination, especially when the product portfolio includes both developmental and commercial manufacturing processes.
Thank you for your interest in our scientific content.
In the ever-evolving landscape of pharmaceutical research, Ardena stands at the forefront with unparalleled expertise in solid state chemistry. Our whitepaper explores the pivotal role of solid state chemistry in drug development, emphasizing how meticulous polymorph screening and rational solid form selection can significantly accelerate the journey from discovery to market. By addressing the complexities and challenges inherent in modern drug development, we ensure stability, bioavailability, and intellectual property protection.
At Ardena, we understand that speed and efficiency are paramount in drug development. Our integrated services and phase-appropriate workflows are tailored to meet the dynamic needs of our clients, from preclinical development to market formulation. Leveraging cutting-edge technologies and maintaining flexible, client-focused collaboration, we help mitigate development risks and streamline the regulatory submission process. Discover how Ardena’s commitment to scientific excellence and strategic outsourcing can transform your drug development program.
Thank you for your interest in our scientific content.
Streamlining Early Drug Development: Efficient Synthesis of Highly Potent Compounds
This whitepaper explores Ardena’s strategic methodology for addressing the challenges in synthesizing highly potent compounds (HPAPI) during early drug development. Emphasizing a rational risk-based approach, the paper outlines key factors in achieving safety, efficiency, and compliance with regulatory standards. The methodology includes continuous risk assessment, facility design based on Occupational Exposure Bands (OEB), and a phased strategy for drug synthesis. With a focus on Quality by Design (QbD) principles, the approach ensures scalability and optimization, allowing for rapid adaptation as more safety data become available. This methodology positions organizations for success in drug development, providing an integrated platform for synthesis and manufacturing from pre-clinical to clinical stages.
By Rob Abbenhuis, CHEM Division Manager at Ardena
Drug discovery of newly identified clinical targets is an exciting and dynamic field for scientists working in academia, start-ups and small to large pharmaceutical companies. During design and screening lead compounds are selected based on their optimum receptor interaction demonstrated in further in-vitro preclinical trials as well as in-vivo animal trials. The continuous advance in medicinal chemistry enables the development of highly potent compounds with the required drug-like properties [1,2]. The increasing focus of drug research on the unmet clinical needs in oncology, inflammatory disease and antiviral compounds lead to more compounds with complex pharmacological activity, toxicity, and benefit-risk profiles [3–5]. Several of these compounds are highly potent active pharmaceutical ingredients (HPAPI) with foreseen pharmacological and toxicological activity at low doses. Compounds with novel pharmacological actions also carry a high risk of unknown toxicology or off-target effects due to the limited safety data and lack of class reference compounds. Such drug candidates are considered as HPAPI by default until their toxicology profile is fully established [6]. To progress quickly into the clinical program a phase appropriate synthesis of sufficient drug substance according to the project needs and stage is required. Due to the limited and unknown safety profiles of such compounds, synthesis is performed in specialized facilities and by a dedicated organization.
Drug synthesis challenges in early development
Timely entry into the clinical trial program depends on the synthesis of sufficient drug substance and is known to be a critical milestone. The challenges arise from the fact that drug synthesis has yet to be developed, while at the same time ensuring the protection of workers and the environment [7]. In addition, there are still no standardized definitions for various toxicological terms and no clear guidelines on the classification of new chemical entities into categories and specific handling requirements [8,9]. Consequently, the available data must be evaluated individually for each compound to derive the necessary safety and precautionary measures during manufacture [10]. A rational risk mitigation approach is applied by Ardena to specifically support fast track, priority, accelerated and break-through designation programs in drug synthesis and drug product manufacturing.
Rational risk-based and phase-appropriate approach in drug synthesis
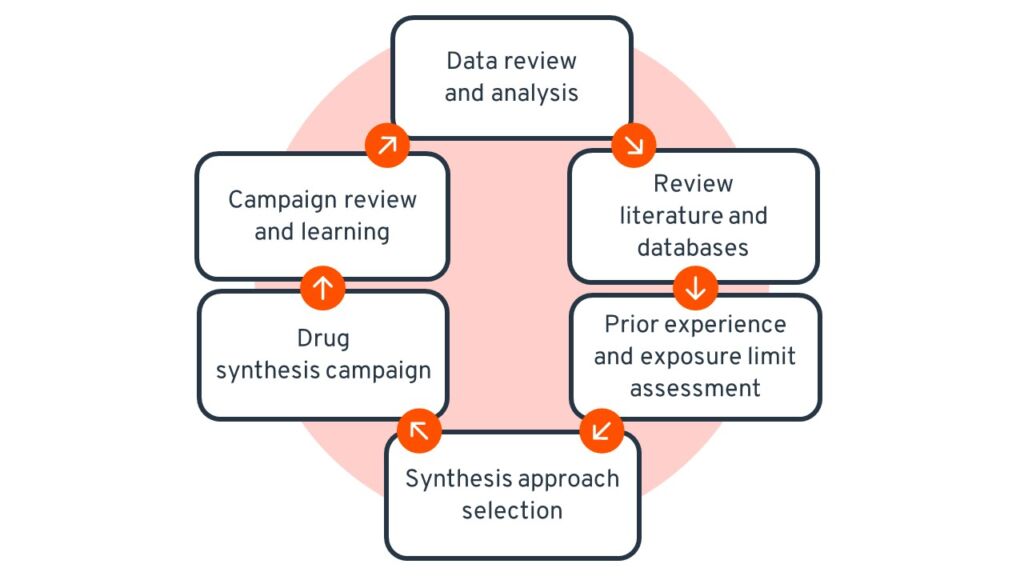
Establishing a comprehensive toxicological profile of a new compound remains difficult in the early stages of drug development and can significantly impact the development program as data generation is a lengthy and expensive process [11,12]. The lack of data, particularly concerning the novel pharmacological actions of compounds potentially designated as Highly Potent Active Pharmaceutical Ingredients (HPAPI), and the transformation of process intermediates to conform with acceptable occupational exposure levels, poses a significant obstacle. To overcome this issue in early drug discovery, a risk-based approach is considered [13,14]. This approach consists of a combination of a comprehensive review of the available safety, preclinical and physicochemical compound properties, a risk assessment including available data bases and literature (e.g., ISPE Risk MaPP [15]; US NIOSH Hazardous drug alert [16], etc.,), layout and design of the synthesis process including facility, equipment, flow and a risk mitigation strategy, conducted before running the first synthesis campaign (Figure 1).
Figure 1: Rational risk mitigation approach and continuous improvement process applied by Ardena.
The risk-based approach will be continuously up-dated as more clinical and safety relevant data become available during the course of the project. The objective continues to be identification of and adaptation to the most efficient and cost-effective API synthesis compliant with the Environment, Health and Safety (EHS) standards and regulatory requirements during the drug development program.
The goal of the initial and follow-up risk assessments is to determine and update the relevant safety values throughout the manufacturing of the drug substance. The Acceptable Daily Exposure (ADE) and Permitted Daily Exposure (PDE), used in Europe and USA respectively, describe the acceptable limits (mg/kg body weight) of cross-contamination of intermediates or drug products due to dust, vapors, aerosols, equipment, or operators clothing etc. during the manufacturing process [17,18]. The Occupational Exposure Limits (OEL) describe the acceptable limits of specific airborne particles (µg/m3) to which workers may be exposed, as well as the type of exposure risks such as inhalation or dermal [19]. While the ADE/PDE and OEL levels are established upon completion of the full safety data package, Occupational Exposure Bands (OEB) are used during the early development (Figure 2). The OEB classification is based on the initial risk assessment considering an exposure range to the compound at negligible risk for the workers and ensures that efficient precautionary measures are in place during synthesis [20]. The determination of the limits is compound specific and considers if the compound is toxic at any value (e.g., cytotoxic, genotoxic) or has a threshold value of toxicity (e.g., No-Observed-Adverse-Effect Level (NOAEL)) [21]
Figure 2: OEB banding strategy used by Ardena assuring safety and efficiency in drug synthesis at all development stages.
Drug synthesis: Process development and design
After determining the operative OEB for the compound and its intermediates during synthesis, the potential synthetic routes are evaluated and the most suitable synthesis approaches are defined. The selection of the synthesis processes to be evaluated will be guided by the principles of efficiency, low exposure risk, scalability, and analytical monitoring for example by qualified process analytical technologies.
Based on experience and knowledge a parallel synthesis approach is applied to accelerate the development according to Quality by Design (QbD) principles [22]. Potentially Critical Quality Attributes (CQA) are identified early in the process to establish the design space and determine the safest and most efficient synthesis. Building a solid understanding of the operational parameter ranges supports process optimization, scaling up, robustness, consistent quality, and safety of the synthesis. Other factors considered include facility lay out, equipment design, personnel, process flow, cleaning, and desired physical API properties most suitable for the finished product development and manufacturing.
Alongside the synthetic process, sensitive qualitative and quantitative analytical methods for the intermediates, artefacts, impurities, and the compound are developed and qualified. The analytics are essential for the safety and cleaning monitoring as well as the process monitoring even within a contained chemical manufacturing process.
Manufacturing of HPAPI and compounds with limited toxicity data
In order to deal with unknowns and uncertainties due to limited toxicology data in compound synthesis each manufacturing campaign is performed under the relevant safety conditions and precautions as well as according to the ICH Q11 guideline [23]. Based on experience of chemists, toxicologists, and high-profile operators familiar and trained on HPAPI manufacturing, a good balance between effective safety, development timelines and investments are established. Hazard analysis and hygiene risk assessments determine if the synthesis or certain steps of the synthesis require a segregation concept, pressure cascading or containment technology. In addition, automated systems can be considered which allow continuous recording of the process parameters to support design space development of CQA through data acquisition, evaluation and analysis.
The qualification of multipurpose process equipment includes a cleanability assessment using swab or rinse analysis and fluorescent dye testing to study as well as establish and qualify the cleaning procedure. Post manufacturing the cleaning process is validated by various verification tests to confirm the absence of any cross-contamination.
The adoption of a risk-based approach to expedite drug synthesis and facilitate the supply of pre-clinical and clinical materials, even in the absence of comprehensive toxicology and safety data, can be optimized through the default adoption of segregation and containment technologies. Gathering important information and experience with the synthesis by the above approach allows rapid optimization, simplification, and classification into the appropriate OEB band as further safety data arise and the full safety data package will become available before the commercialization of the product.
Conclusion
The exponential advances in biomedical sciences have shifted drug discovery towards unmet medical needs and substantially shorter drug development timelines [24]. Such programs face the challenge of the need for drug substance synthesis with only limited toxicological and pharmacological data or predicted HPAPI classification. Ardena has extensive expertise in finding the right balance between EHS, regulatory requirements, early entry into clinical trials and rapid patient access. Dedicated manufacturing and analytical facilities, experienced and trained workers, and established rational risk-based approaches provide an integrated platform for synthesis and drug product manufacturing at the critical interface from the pre-clinical through to the clinical stage. The approach follows a long-term perspective by accruing product and process knowledge and controls based on QbD principles to ensure scalability and transferability as well as manufacturing optimization and simplification as more safety data become available.
[1] A.G. Schwaid, I. Cornella-Taracido, Causes and Significance of Increased Compound Potency in Cellular or Physiological Contexts, J Med Chem. 61 (2018) 1767–1773. https://doi.org/10.1021/acs.jmedchem.7b00762.
[2] D.R. Owen, C.M. N Allerton, A.S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R.D. Cardin, A. Carlo, K.J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K.S. Gajiwala, S.A. Gibson, S.E. Greasley, B.L. Hurst, E.P. Kadar, A.S. Kalgutkar, J.C. Lee, J. Lee, W. Liu, S.W. Mason, S. Noell, J.J. Novak, R. Scott Obach, K. Ogilvie, N.C. Patel, M. Pettersson, D.K. Rai, M.R. Reese, M.F. Sammons, J.G. Sathish, R.P. Shankar Singh, C.M. Steppan, A.E. Stewart, J.B. Tuttle, L. Updyke, P.R. Verhoest, L. Wei, Q. Yang, Y. Zhu, An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19, n.d. https://www.science.org.
[3] C.A. Pinto, Z. Balantac, S. Mt-Isa, X. Liu, O.L. Bracco, H. Clarke, T. Tervonen, Regulatory benefit–risk assessment of oncology drugs: A systematic review of FDA and EMA approvals, Drug Discov Today. 28 (2023). https://doi.org/10.1016/j.drudis.2023.103719.
[4] C. Moreau Bachelard, E. Coquan, P. du Rusquec, X. Paoletti, C. Le Tourneau, Risks and benefits of anticancer drugs in advanced cancer patients: A systematic review and meta-analysis, EClinicalMedicine. 40 (2021). https://doi.org/10.1016/j.eclinm.2021.101130.
[5] M. Senior, Fresh from the biotech pipeline: fewer approvals, but biologics gain share, Nat Biotechnol. 41 (2023) 174–182. https://doi.org/10.1038/s41587-022-01630-6.
[6] R.G. Sussman, A.R. Schatz, T.A. Kimmel, A. Ader, B.D. Naumann, P.A. Weideman, Identifying and assessing highly hazardous drugs within quality risk management programs, Regulatory Toxicology and Pharmacology. 79 (2016) S11–S18. https://doi.org/10.1016/j.yrtph.2016.05.025.
[7] D.C. Blakemore, L. Castro, I. Churcher, D.C. Rees, A.W. Thomas, D.M. Wilson, A. Wood, Organic synthesis provides opportunities to transform drug discovery, Nat Chem. 10 (2018) 383–394. https://doi.org/10.1038/s41557-018-0021-z.
[8] R.G. Sussman, B.D. Naumann, T. Pfister, C. Sehner, C. Seaman, P.A. Weideman, A harmonization effort for acceptable daily exposure derivation – Considerations for application of adjustment factors, Regulatory Toxicology and Pharmacology. 79 (2016) S57–S66. https://doi.org/10.1016/j.yrtph.2016.05.023.
[9] P.A. Weideman, A.M. Pecquet, M.A. Maier, Harmonization efforts for deriving health-based exposure limits in the pharmaceutical industry – Advancing the current science and practice, Regulatory Toxicology and Pharmacology. 79 (2016) S1–S2. https://doi.org/10.1016/j.yrtph.2016.07.016.
[10] J. Gould, C.M. Callis, D.G. Dolan, B. Stanard, P.A. Weideman, Special endpoint and product specific considerations in pharmaceutical acceptable daily exposure derivation, Regulatory Toxicology and Pharmacology. 79 (2016) S79–S93. https://doi.org/10.1016/j.yrtph.2016.05.022.
[11] E.C. Faria, J.P. Bercu, D.G. Dolan, E.J. Morinello, A.M. Pecquet, C. Seaman, C. Sehner, P.A. Weideman, Using default methodologies to derive an acceptable daily exposure (ADE), Regulatory Toxicology and Pharmacology. 79 (2016) S28–S38. https://doi.org/10.1016/j.yrtph.2016.05.026.
[12] T. Janela, J. Bajorath, Large-Scale Predictions of Compound Potency with Original and Modified Activity Classes Reveal General Prediction Characteristics and Intrinsic Limitations of Conventional Benchmarking Calculations, Pharmaceuticals. 16 (2023). https://doi.org/10.3390/ph16040530.
[13] F. Pognan, M. Beilmann, H.C.M. Boonen, A. Czich, G. Dear, P. Hewitt, T. Mow, T. Oinonen, A. Roth, T. Steger-Hartmann, J.P. Valentin, F. Van Goethem, R.J. Weaver, P. Newham, The evolving role of investigative toxicology in the pharmaceutical industry, Nat Rev Drug Discov. 22 (2023) 317–335. https://doi.org/10.1038/s41573-022-00633-x.
[14] E.A.G. Blomme, Y. Will, Toxicology Strategies for Drug Discovery: Present and Future, Chem Res Toxicol. 29 (2016) 473–504. https://doi.org/10.1021/acs.chemrestox.5b00407.
[15] ISPE, ISPE Risk MaPP, (2017).
[16] FDA, NIOSH Hazardous Drug Alert, (2023).
[17] EMA, Committee for Medicinal Products for Human Use (CHMP) Committee for Medicinal Products for Veterinary Use (CVMP) Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities, 2014. www.ema.europa.eu/contact.
[18] E. V. Sargent, E. Faria, T. Pfister, R.G. Sussman, Guidance on the establishment of acceptable daily exposure limits (ADE) to support Risk-Based Manufacture of Pharmaceutical Products, Regulatory Toxicology and Pharmacology. 65 (2013) 242–250. https://doi.org/10.1016/j.yrtph.2012.12.007.
[19] C. Jandard, H. Hemming, M. Prause, C. Sehner, M. Schwind, M. Abromovitz, E. Lovsin Barle, Applicability of surface sampling and calculation of surface limits for pharmaceutical drug substances for occupational health purposes, Regulatory Toxicology and Pharmacology. 95 (2018) 434–441. https://doi.org/10.1016/j.yrtph.2017.12.020.
[20] J.C. Graham, J. Hillegass, G. Schulze, Considerations for setting occupational exposure limits for novel pharmaceutical modalities, Regulatory Toxicology and Pharmacology. 118 (2020). https://doi.org/10.1016/j.yrtph.2020.104813.
[21] D.G. Dolan, B.D. Naumann, E. V. Sargent, A. Maier, M. Dourson, Application of the threshold of toxicological concern concept to pharmaceutical manufacturing operations, Regulatory Toxicology and Pharmacology. 43 (2005) 1–9. https://doi.org/10.1016/j.yrtph.2005.06.010.
[22] ICH Q8R2, Committee for Human Medicinal Products ICH guideline Q8 (R2) on pharmaceutical development, 2017. www.ema.europa.eu/contact.
[23] ICH Q11, ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/ biological entities), 2012. www.ema.europa.eu.
[24] FDA, 2022 Annual Report, 2023.
Exploring the Science and Technology Behind Fractionation, Purification, and Downstream Processing: From Discovery to Commercialization
With increasing progress in the life sciences, we are constantly broadening our ability to treat diseases that were previously incurable. This progress however comes with new challenges due to the growing complexity of chemical and biotherapeutic products. Developing and manufacturing the compound of interest in a pure and stable form quickly and efficiently is considered to be business critical if not the decisive factor for success.
The growing complexity of chemical synthesis and biotechnological expression systems is associated with a need for more intensive fractionation and purification downstream processes to ensure sufficient product quality. Increasingly stringent quality criteria for purity are required to secure efficacy, potency, and stability, and prevent toxicity and immunogenicity, especially for biological products such as proteins, vaccines, and monoclonal antibodies. This involves the elimination of any impurities or by-products to achieve the desired compound purity while maintaining the chemical or biological stability and activity in a consistent manner.
The technical challenge of the specificity required for each product and process has led to a tremendous effort to better understand downstream processes and make them more efficient. The more technological options and processes that are available and offered, the more important the expertise in selecting, developing and scaling the most efficient approach from these options becomes.
With increasing progress in the life sciences, we are constantly broadening our ability to treat diseases that were previously incurable. This progress however comes with new challenges due to the growing complexity of chemical and biotherapeutic products. Developing and manufacturing the compound of interest in a pure and stable form quickly and efficiently is considered to be business critical if not the decisive factor for success.
The growing complexity of chemical synthesis and biotechnological expression systems is associated with a need for more intensive fractionation and purification downstream processes to ensure sufficient product quality. Increasingly stringent quality criteria for purity are required to secure efficacy, potency, and stability, and prevent toxicity and immunogenicity, especially for biological products such as proteins, vaccines, and monoclonal antibodies. This involves the elimination of any impurities or by-products to achieve the desired compound purity while maintaining the chemical or biological stability and activity in a consistent manner.
The technical challenge of the specificity required for each product and process has led to a tremendous effort to better understand downstream processes and make them more efficient. The more technological options and processes that are available and offered, the more important the expertise in selecting, developing and scaling the most efficient approach from these options becomes.
The goal is to retrieve sufficiently pure chemical and biological compounds required for the development stage, including special reagents for binding assays [read our whitepaper on bioanalysis of ADCs here] as well as GMP grade material be it an adjuvant or an active compound to support the clinical trials and commercialization. With the downstream processing contributing between 50 – 80% to the compound manufacturing costs, increasing the efficiency and simplifying the downstream process has become another important goal for drug development (Labrou, 2014).
Principles of separation and purification
The upstream processing of the chemical synthesis or biotechnology expression systems (plants, cellular, etc) is an important and critical step in the manufacture of the specific compound. The process parameters applied determine the levels and types of impurities or by-products present after the upstream step in the compound-containing substrate. The upstream process thus determines the complexity and number of fractionation and purification steps required for the downstream process.
The separation and purification of a compound is typically achieved through a variety of different physical and chemical properties which are utilized to separate the compound of interest from impurities and by-products. These compound-specific properties include size, shape, charge, isoelectric point, charge distribution, hydrophobicity, solubility, density, ligand-binding affinity, metal binding, reversible association, posttranslational modifications, and specific sequences or structures (Labrou, 2014). The major technologies used are filtration, chromatography, extraction, precipitation, and centrifugation. Filtration, extraction, precipitation, and centrifugation are mainly used for harvesting the compound-containing fraction from the up-scaling, while chromatographic and advanced filtration technologies are used for the final purification step. For complex mixtures, in particular, a scheme of different purification steps is necessary to eliminate impurities and by-products step by step to retrieve the pure compound.
Chromatography is a versatile purification technique, separating the compounds based on their size, physico-chemical, or hydrophobic/hydrophilic properties (see Table 1).
Chromatographic purification principles
Affinity (AFC)
Separation method based on a specific binding interaction between an immobilized ligand and its binding partner
Ion exchange (CEX, AEX)
Separation based on ions and charged molecules by affinity to the ion exchanger
Hydrophobic interaction (HIC)
Separation of molecules according to differences in their surface hydrophobicity
Size exclusion (SEC)
Separation of molecules based on their size by filtration through a gel
Reverse phase (RPC)
Separation by a hydrophobic stationary phase and a polar mobile phase
Multimodal (MMC)
Separation by more than one form of interaction between the stationary phase and analytes
Table 1: Main chromatographic purification technologies and their purification principles
Purification by chromatography is a well understood technology, however it requires substantial expertise and knowledge as well as selective and sensitive analytical capabilities to successfully evaluate the impact of potentially critical variables (Hanke & Ottens, 2014).
Filtration methods generally consist of surface filtration, depth filtration, cross-filtration, and ultra-filtration technologies. Surface filters are single layer filters defined by their pore size. Depth filters are porous matrices purifying effectively throughout the entire depth and their efficiency can be further increased through agitation. Cross-filtration technologies such as Tangential Flow Filtration (TFF) leverage a high velocity flow tangentially across the membrane surface to purify proteins or nanoparticles. Diafiltration (DF) is a cross-flow filtration technique in which fresh solvent is added to replace the volume loss due to the filtration process. Ultra-Filtration (UF) or Nano-Filtration (NF) are pressure-driven membrane transport technologies passing mixtures through hollow fibers of membrane material. Due to the tangential filtration mode, cross-flow filtration technology prevents filter clogging or fouling which is especially important for the purification of nanoparticles (Busatto et al., 2018). This also applies to the development and commercial manufacturing of new vaccine and vaccine adjuvant nanoparticle products (Pires et al., 2023).
Approaches in the development of fractionation and purification towards downstream processing
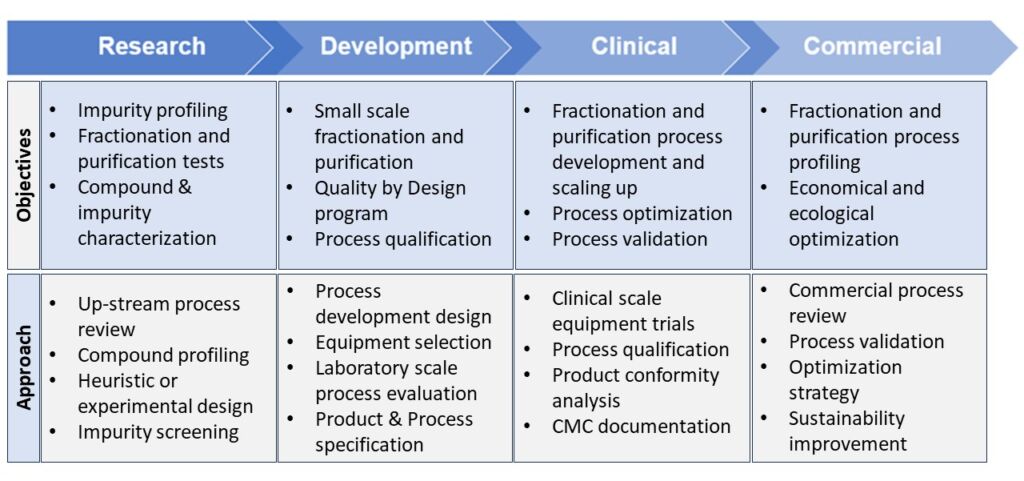
A systematic yet practical approach for fractionation and purification is required when starting the process development to achieve the desired objectives and milestones in each phase. Table 2 shows some important objectives of fractionation and purification at each stage and demonstrates that fractionation and purification should start at the drug discovery phase and continue through to a validated and commercially viable downstream process.
Table 2: Objectives and approaches for fractionation and purification throughout the drug discovery and development phases and seamless transition into the commercial downstream process
For each compound, be it derived from chemical synthesis, biotechnological expression system, or natural origin, a rational and product specific development strategy is required. After reviewing the synthesis or expression and product characteristics a universal or process specific heuristic approach can be used effectively for known products and processes (Pfister et al., 2017). In case of new products and processes, an experimental approach may be more target-oriented and can be performed in a high throughput manner. These high throughput process development (HTPD) tools consist of parallel, miniaturized automated systems which can be used for the upstreaming and downstreaming of the product development (Baumann & Hubbuch, 2017) .
Standard or platform approaches for purification have also been suggested for monoclonal antibodies, which require only limited development work and have a high probability to achieve the degree of purity required (Shukla et al., 2007). While they might serve the purpose during the development stage, they might not provide the most efficient option for the commercial scale due to the expensive Protein A chromatography step. To overcome this issue techniques like aqueous two-phase separation technique are under investigation (Kruse et al., 2020).
The need to increase efficiency in separation and purification
In the early phases of a development program, especially when entering into a phase 1 clinical trial, downstream processing is time-critical and the speed of its development is a top priority. However, in the later development phase the success-critical factors of the efficiency and scalability of the process come to the fore.
A heuristic approach coupled with HTPD technology based on small scale automatic dispensing and sampling equipment have proven to be extremely powerful in accelerating downstream development in the early phase. For example, preparative high-performance liquid chromatography (HPLC) has evolved as a scalable purification and polishing technology throughout the product life cycle (De Luca et al., 2021). HTPD technology can also be used to generate large sets of experimental data to support development according to the quality-by-design (QbD) principles or a statistical and predictive model-based approach (Keulen et al., 2022). In order to overcome some of the limitations of the purification of peptides by chromatography catch and release (C&R) methods based on a specific combination of base-labile cleavable linkers with oxime-based and hydrazone-based ligation chemistry are under evaluation but these require more work to further reduce off-target reactions (Reimann et al., 2019).
The growing public awareness of climatic change and environmental protection has put pressure on the pharmaceutical industry, the manufacturing processes and products. Sustainability is therefore not only a question of corporate identity but also of a company’s profitability. Sustainability in biopharmaceutical manufacturing must be considered across the entire process, with the downstream processing playing a special role. Improvements can be achieved by using less critical solvents, whereas optimization of processes and procedures can achieve solvent savings of up to 90% (Ferrazzano et al., 2022). These savings are achievable because batch-based processes are typically affected by a yield-purity trade-off, due to overlapping regions where either some the by-products will be carried over or product will be lost due to the narrow fractionation or purification window (De Luca et al., 2020; Ferrazzano et al., 2022). With the technological advances in downstream processing and the use of QbD principles, predictive models can be used to establish a PAT-based close-loop control (Gerzon et al., 2022) which increasingly enables the conversion of the upstream and the downstream processes into a continuous process (Behere & Yoon, 2020). It is estimated that continuous bioprocessing could save approximately 55% of the costs compared to traditional batch manufacturing (Walther et al., 2015). Continuous processing is supported and promoted by the FDA through the introduction of guidelines to improve manufacturing and patient access to innovative biotherapeutics (Fisher et al., 2019).
While the development of a continuous manufacturing process for clinical phases 1 and 2 is disproportionate, individual downstream process steps can already be carried out continuously or later transferred into continuous processing mode. Cross-flow filtration technologies such as TFF, DF, UF and NF are process technologies serving small and large scale as well as batch and continuous processing options (David et al., 2020). Besides their efficient purification these technologies are also suited to producing higher concentrations of biochemical compounds as well as performing the essential buffer changes at the end of the purification step (De Luca et al., 2020).
Enhancing compound concentration and stability can be achieved by eliminating reactive or catalytic impurities through purification, which can be further optimized through an additional lyophilization process. This may be especially important for overcoming the transport and storage issues associated with vaccines for tropical diseases or third world use e.g. Covid vaccines (Crommelin et al., 2021).
Case studies
Ardena has carried out several upstream and downstream programs in partnership with small and large enterprises to support chemical and biological development programs from the very early phases through to commercial scale.
In a phase-appropriate development approach, especially in early phases when the demand for drug substance is still limited, the common drawbacks of chromatographic methods such as cost and throughput may be offset by benefits: time of delivery and purity. In critical cases where the impurity profile is challenging and development of the chemical process is time consuming, the application of chromatographic methods provides a clear advantage.
In one of our projects a key starting material for GMP manufacturing was prepared via a photo-isomerization reaction on a steroid substrate which produced a mixture of the target compound together with a range of impurities. The mixture contained about 50% of the desired material hence crystallization was not feasible. Gravity chromatography was developed and scaled which allowed the production of the material on a multikilogram scale.
With the successful launch of Covid vaccines mRNA delivery technology has become one of the most exciting and promising approaches for innovative therapies. For the stabilization and transfection of the mRNA, specifically designed ionic lipids or lipoids are an essential part of the delivery system (Zhang et al., 2021). Using an innovative cationic lipidoid derivative identified by a company in a screening program, a complex ionic lipoid was custom-synthesized and more than 500 g of GMP grade material was produced. Upscaling required the development of an upstream synthesis route and a downstream purification process that would meet the efficiency requirements of the commercial manufacturing process. The development of the synthesis was oriented towards high selectivity and avoidance of impurities that are difficult to separate. Through a purification step of an intermediate in the synthesis by using reverse phase chromatography, the upstream product was obtained with high purity. For the final downstream processing, flash chromatography was successfully applied together with preparative HPLC coupled to UV-Vis and Evaporative Light Scattering detectors. The equipment used to process 500 g of GMP grade of the ionic lipoid, a Buchi C850 system, facilitated both rapid production of sufficient quantities for subsequent clinical development and seamless transfer to larger-scale equipment for commercial volumes.
Fractionation and purification of sensitive nanoparticles (e.g. vaccines, liposomes etc.) is a growing field of advanced drug delivery. One example is a new vaccine adjuvant which was developed by one of our customers, targeting the immune stimulatory complexes (ISCOMs) to increase the immune response of protein-based vaccines. This matrix is based on saponins of natural origin. An extract is fractionated and the fractions are collected and lyophilized. The fractions are formulated with phospholipids and cholesterol to retrieve two distinct nanoparticles (Bengtsson et al., 2011; Magnusson et al., 2018).
During a fast-track development project by a major healthcare company, purification of an innovative Manganese-based contrast agent became the bottleneck in development and threatened to significantly delay entry into clinical development. Following the upstream process, the compound containing aqueous solution was passed through a nanofiltration TFF process to separate molecules with a molecular weight of 100-250 Da from the compound. Ion-exchange chromatography was selected due to the affinity profile of the compound and a stationary phase, mobile phase, and gradient were developed to retrieve high yields of pure GMP grade compound from the pre-purified solution. Due to the instability of the compound in solution, a lyophilization step was added to provide the GMP grade compound in a stable solid form. The downstream process developed leveraged prior experience on a volume-efficient and scalable approach toward commercial manufacturing.
Conclusion
Purification plays an important role at multiple stages of drug development and manufacturing of complex drug synthesis and biotechnology expression systems. Developing an effective and efficient downstream purification process is upstream and product dependent and requires individually tailored purification schemes.
It is therefore important to choose the right approach for the upstream and downstream processes that ensures safe, reproducible purification and can be efficiently transferred to a manufacturing scale. It is important to use established processes whenever possible and to avoid complex and lengthy purification procedures and overengineering.
The challenge of considering commercial manufacturing early in the development phase is often underestimated. The difficulty arises from the wide variety of parameters and equipment variables which are being tested during development without consideration for the readiness of the future commercial manufacturing facility (Chahar et al., 2020; Matte, 2022).
Science and knowledge-based approaches coupled with strong analytical capabilities accelerate the time critical downstream process development including the lay out towards the most efficient commercial scale purification. Depending on their internal infrastructure and organizational setup, companies can greatly benefit from outsourcing development and manufacturing processes.
References
Baumann, P., & Hubbuch, J. (2017). Downstream process development strategies for effective bioprocesses: Trends, progress, and combinatorial approaches. In Engineering in Life Sciences (Vol. 17, Issue 11, pp. 1142–1158). Wiley-VCH Verlag. https://doi.org/10.1002/elsc.201600033
Behere, K., & Yoon, S. (2020). Chromatography bioseparation technologies and in-silico modelings for continuous production of biotherapeutics. Journal of Chromatography A, 1627. https://doi.org/10.1016/j.chroma.2020.461376
Busatto, S., Vilanilam, G., Ticer, T., Lin, W. L., Dickson, D. W., Shapiro, S., Bergese, P., & Wolfram, J. (2018). Tangential flow filtration for highly efficient concentration of extracellular vesicles from large volumes of fluid. Cells, 7(12). https://doi.org/10.3390/cells7120273
Chahar, D. S., Ravindran, S., & Pisal, S. S. (2020). Monoclonal antibody purification and its progression to commercial scale. In Biologicals (Vol. 63, pp. 1–13). Academic Press. https://doi.org/10.1016/j.biologicals.2019.09.007
Crommelin, D. J. A., Anchordoquy, T. J., Volkin, D. B., Jiskoot, W., & Mastrobattista, E. (2021). Addressing the Cold Reality of mRNA Vaccine Stability. Journal of Pharmaceutical Sciences, 110(3), 997–1001. https://doi.org/10.1016/j.xphs.2020.12.006
David, L., Schwan, P., Lobedann, M., Borchert, S. O., Budde, B., Temming, M., Kuerschner, M., Alberti Aguilo, F. M., Baumarth, K., Thüte, T., Maiser, B., Blank, A., Kistler, V., Weber, N., Brandt, H., Poggel, M., Kaiser, K., Geisen, K., Oehme, F., & Schembecker, G. (2020). Side-by-side comparability of batch and continuous downstream for the production of monoclonal antibodies. Biotechnology and Bioengineering, 117(4), 1024–1036. https://doi.org/10.1002/bit.27267
De Luca, C., Felletti, S., Lievore, G., Chenet, T., Morbidelli, M., Sponchioni, M., Cavazzini, A., & Catani, M. (2020). Modern trends in downstream processing of biotherapeutics through continuous chromatography: The potential of Multicolumn Countercurrent Solvent Gradient Purification. In TrAC – Trends in Analytical Chemistry (Vol. 132). Elsevier B.V. https://doi.org/10.1016/j.trac.2020.116051
De Luca, C., Lievore, G., Bozza, D., Buratti, A., Cavazzini, A., Ricci, A., Macis, M., Cabri, W., Felletti, S., & Catani, M. (2021). Downstream processing of therapeutic peptides by means of preparative liquid chromatography. In Molecules (Vol. 26, Issue 15). MDPI AG. https://doi.org/10.3390/molecules26154688
Ferrazzano, L., Catani, M., Cavazzini, A., Martelli, G., Corbisiero, D., Cantelmi, P., Fantoni, T., Mattellone, A., De Luca, C., Felletti, S., Cabri, W., & Tolomelli, A. (2022). Sustainability in peptide chemistry: Current synthesis and purification technologies and future challenges. In Green Chemistry (Vol. 24, Issue 3, pp. 975–1020). Royal Society of Chemistry. https://doi.org/10.1039/d1gc04387k
Fisher, A. C., Kamga, M. H., Agarabi, C., Brorson, K., Lee, S. L., & Yoon, S. (2019). The Current Scientific and Regulatory Landscape in Advancing Integrated Continuous Biopharmaceutical Manufacturing. In Trends in Biotechnology (Vol. 37, Issue 3, pp. 253–267). Elsevier Ltd. https://doi.org/10.1016/j.tibtech.2018.08.008
Gerzon, G., Sheng, Y., & Kirkitadze, M. (2022). Process Analytical Technologies – Advances in bioprocess integration and future perspectives. Journal of Pharmaceutical and Biomedical Analysis, 207. https://doi.org/10.1016/j.jpba.2021.114379
Hanke, A. T., & Ottens, M. (2014). Purifying biopharmaceuticals: Knowledge-based chromatographic process development. In Trends in Biotechnology (Vol. 32, Issue 4, pp. 210–220). Elsevier Ltd. https://doi.org/10.1016/j.tibtech.2014.02.001
Keulen, D., Geldhof, G., Bussy, O. Le, Pabst, M., & Ottens, M. (2022). Recent advances to accelerate purification process development: A review with a focus on vaccines. Journal of Chromatography A, 1676. https://doi.org/10.1016/j.chroma.2022.463195
Kruse, T., Kampmann, M., Rüddel, I., & Greller, G. (2020). An alternative downstream process based on aqueous two-phase extraction for the purification of monoclonal antibodies. Biochemical Engineering Journal, 161. https://doi.org/10.1016/j.bej.2020.107703
Labrou, N. E. (2014). Protein Purification: An overview. In N. E. Labrou (Ed.), Methods in Molecular Biology: Protein Downstream Processing (Vol. 1129, pp. 3–10). http://www.springer.com/series/7651
Lövgren Bengtsson, K., Morein, B., & Osterhaus, A. D. (2011). ISCOM technology-based Matrix MTM adjuvant: Success in future vaccines relies on formulation. In Expert Review of Vaccines (Vol. 10, Issue 4, pp. 401–403). https://doi.org/10.1586/erv.11.25
Magnusson, S. E., Altenburg, A. F., Bengtsson, K. L., Bosman, F., de Vries, R. D., Rimmelzwaan, G. F., & Stertman, L. (2018). Matrix-MTM adjuvant enhances immunogenicity of both protein- and modified vaccinia virus Ankara-based influenza vaccines in mice. Immunologic Research, 66(2), 224–233. https://doi.org/10.1007/s12026-018-8991-x
Matte, A. (2022). Recent Advances and Future Directions in Downstream Processing of Therapeutic Antibodies. In International Journal of Molecular Sciences (Vol. 23, Issue 15). MDPI. https://doi.org/10.3390/ijms23158663
Pfister, D., David, L., Holzer, M., & Nicoud, R. M. (2017). Designing affinity chromatographic processes for the capture of antibodies. Part I: A simplified approach. Journal of Chromatography A, 1494, 27–39. https://doi.org/10.1016/j.chroma.2017.02.070
Pires, I. S., Ni, K., Melo, M. B., Li, N., Ben-Akiva, E., Maiorino, L., Dye, J., Rodrigues, K. A., Yun, D. S., Kim, B., Hosn, R. R., Hammond, P. T., & Irvine, D. J. (2023). Controlled lipid self-assembly for scalable manufacturing of next-generation immune stimulating complexes. Chemical Engineering Journal, 464. https://doi.org/10.1016/j.cej.2023.142664
Reimann, O., Seitz, O., Sarma, D., & Zitterbart, R. (2019). A traceless catch-and-release method for rapid peptide purification. Journal of Peptide Science, 25(1). https://doi.org/10.1002/psc.3136
Shukla, A. A., Hubbard, B., Tressel, T., Guhan, S., & Low, D. (2007). Downstream processing of monoclonal antibodies-Application of platform approaches. In Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences (Vol. 848, Issue 1, pp. 28–39). https://doi.org/10.1016/j.jchromb.2006.09.026
Walther, J., Godawat, R., Hwang, C., Abe, Y., Sinclair, A., & Konstantinov, K. (2015). The business impact of an integrated continuous biomanufacturing platform for recombinant protein production. Journal of Biotechnology, 213, 3–12. https://doi.org/10.1016/j.jbiotec.2015.05.010
Zhang, Y., Sun, C., Wang, C., Jankovic, K. E., & Dong, Y. (2021). Lipids and Lipid Derivatives for RNA Delivery. In Chemical Reviews (Vol. 121, Issue 20, pp. 12181–12277). American Chemical Society. https://doi.org/10.1021/acs.chemrev.1c00244
Phase-appropriate analytical method development
Drug development requires an average overall investment of USD 1.5 – 4.5 billion and remains at high risk due to the huge attrition rate phase after phase. The pre-clinical development phase and the phase 1 clinical trial show the highest attrition rate due to toxicity or drug metabolism and pharmacokinetic issues. Mitigating the risk during a new drug candidate development program starting from the discovery phase onwards remains essential for drug development efficiency and success.
The challenge is to perform the necessary steps in a structured and predictive manner which is also tailored to the project at hand. This can be achieved by committing the resources to stage-appropriate steps and confirming their suitability for the following steps.
This structured and stepwise project plan reduces the level of investments into future work prior to receiving positive results from the earlier stage which would justify such investments.
Doing the right things at the right time
Drug development requires an average overall investment of USD 1.5 – 4.5 billion and remains at high risk due to the huge attrition rate phase after phase [1; 2]. The pre-clinical development phase and the phase 1 clinical trial show the highest attrition rate due to toxicity or drug metabolism and pharmacokinetic issues [3; 4]. Mitigating the risk during a new drug candidate development program starting from the discovery phase onwards remains essential for drug development efficiency and success.
The challenge is to perform the necessary steps in a structured and predictive manner which is also tailored to the project at hand. This can be achieved by committing the resources to stage-appropriate steps and confirming their suitability for the following steps. This structured and stepwise project plan reduces the level of investments into future work prior to receiving positive results from the earlier stage which would justify such investments.
For small molecules and biologics, development of the compound synthesis including analytical development is the starting point into the pre-clinical and clinical program. Drug synthesis for toxicology and animal studies is a critical development step as it also serves the clinical trial program. The evaluation of toxicity depends on the chemical structure of the active compound and the purity achieved in the drug synthesis. The route of synthesis, the raw materials and solvents used, as well as the process conditions applied are important factors for the potential formation of harmful impurities. To assure the validity of the outcomes of the toxicology study, a robust and efficient drug synthesis including reliable analytical procedures is essential as this provides the necessary consistency throughout the program.
Developing a robust drug substance synthesis
Setting the compound profile targets and performing an intensive risk assessment using a structured approach, as well as prior knowledge and compound synthesis expertise ensure that the resulting material fulfills the defined quality criteria. Developing the drug substance synthesis has to be accompanied by a thorough understanding of the physicochemical properties and their relevance for drug development [5]. For example, chemical structures that are prone to drug instability or degradation, residual solvents, or formation of chemical artefacts might have a negative impact on the toxicology profile that would be preventable [6]. Other critical factors like salt selection, druggability, solubility, BA, or potential risk for polymorphs, etc. might also arise from the risk assessment and can be addressed. The expected outcome is the definition of potential Critical Material Attributes (CMA) and Critical Quality Attributes (CQA), summarized in preliminary/target specifications, at early stages in order to prevent issues with the compound and its synthesis [7; 8]. Drug substance synthesis must therefore be accompanied by scientifically sound, product-specific analytical procedures for the characterization of the active and residual byproducts. The analytical methods will establish a comprehensive drug substance profile which will be the benchmark for the clinical and commercial phase.
Developing phase-appropriate analytical methods for the active compound
The analytical method development during early-stage drug development is an evolving process that builds successively on sound scientific approaches and data as described by the FDA (Box 1).
21 CFR 312.23 Investigational new drug application (IND)
7 (i) … FDA recognizes that modifications to the method of preparation of the new drug substance and dosage form and changes in the dosage form itself are likely as the investigation progresses. Therefore, the emphasis in an initial Phase 1 submission should generally be placed on the identification and control of the raw materials and the new drug substance. Final specifications for the drug substance and drug product are not expected until the end of the investigational process.
7 (iv) (a ) Drug substance. A description of the drug substance, including its physical, chemical, or biological characteristics; the name and address of its manufacturer; the general method of preparation of the drug substance; the acceptable limits and analytical methods used to assure the identity, strength, quality, and purity of the drug substance; and information sufficient to support stability of the drug substance during the toxicological studies and the planned clinical studies. Reference to the current edition of the United States Pharmacopeia – National Formulary may satisfy relevant requirements in this paragraph.
Box 1: Requirements for drug substance [15]
Regulatory authorities consider this as phase or stage-appropriate analytical method development. They expect only the full analytical method validation according to ICH guidelines to be provided along with the final drug product presentation entering into phase 3 clinical trials. During the pre-clinical and IND phase regulatory authorities request that “an analytical procedure is to demonstrate that it is suitable for its intended purpose” [9]. This is further specified in the ICH Q7 guideline by stating that “while analytical methods performed to evaluate a batch of API for clinical trials may not yet be validated, they should be scientifically sound.” [10]. The selection and development of a scientifically sound analytical method requires experience and understanding of a method principle and capability to measure the targeted CMQ or CQA reliably. For example, to determine elemental impurities Inductively Coupled Plasma Mass Spectroscopy (ICP-MS) or for residual solvents
Head-space Gas Chromatography (HSGC) are suitable method principles. The suitability for the method’s intended purpose is often established by validation of a subset of ICH validation parameters.
The analytical development for a drug substance starts with a clear definition of the targeted drug substance profile and the limits of acceptability (table 1). Analytical methods have to be scientifically sound and fit for purpose to accurately measure the target in the specific drug compound.
Table 1: Ardena’s targeted drug substance profile of quality attributes, acceptance criteria and potential analytical procedures for GLP toxicology study use
(*) In this early phase a well-characterized analytical reference standard may not be available, hence assay by HPLC cannot be used.
Phase-appropriate analytical method development in the scope of product development
The pharmaceutical development process consists of a matrix of different work steps, investigations, and documentation, all of which are interrelated and build on each other (Figure 1). A successful development plan for a new drug product consists of a systematic and knowledge-based approach. This is especially true for the synthesis development of the drug substance, which enables the production of a highly pure drug substance in a reproducible and scalable manner. During clinical stages analytical methods for qualification of the raw materials and intermediates of the synthesis are established as well. Critical solvents and process parameters should be kept to a minimum. The ultimate objective is to manufacture the drug substance by the same synthetic route, and purification and crystallization processes from the toxicology supplies through to the market. In practice however, continuous improvements are often encountered up to the start of process validation. From the beginning the critical equipment and ancillary systems used in manufacturing and control must be suitable and qualified for the intended purpose. To streamline the development times and prevent unnecessary stringent API quality (and its associated cost), Ardena uses the demonstration batch of the drug substance as the first GLP batch to cover the supplies for the toxicology studies within the predefined impurity targets. The early provision of the drug substance synthesis provides continuity within the drug development program, leverages important prior product and process knowledge, and reinforces product specific learning according to the ICH Q 11 recommendations [6].
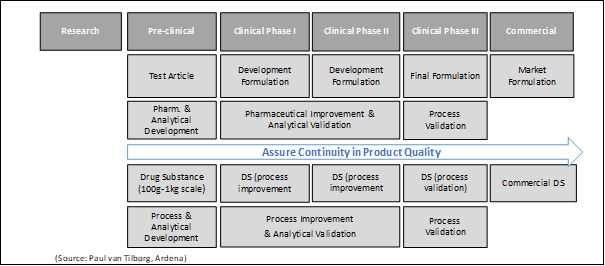
Figure 1: The pharmaceutical drug product and analytical method development process matrix
Early-stage analytical methods development along with the GLP drug substance batch manufacturing will be defined and selected based on a mechanistic understanding of the method principles, internal expertise, and data from prior experiments in accordance with the FDA guidelines [11; 12]. This also ensures that the analytical methods developed are meaningful and can be validated later during the GMP drug substance and drug product manufacturing if the toxicology studies support the nomination of the molecule for further clinical studies. Parameters that can be considered and may be evaluated during analytical method development are specificity, linearity, limits of detection (LOD), limits of quantitation (LOQ), range, accuracy, and precision [10]. The analytical method development in early stage starts with a clearly defined targeted drug substance profile including the quality attributes and acceptance criteria derived from the risk assessment (Table 1). For the toxicology study the goal of the analytical method development is to select the right procedure to determine the individual targets qualitatively and quantitatively with sufficient reliability. This includes the detection of predictable and unpredictable impurities and their characterization [13]. Each individual step and every result must be precisely documented, and corresponding samples have to be taken for later validation purposes. It should be noted that the suitability of the method as well as important validation parameters are revisited and justified during the course of the clinical program based on increasing learning and evidence, and possible changes in the manufacturing process. It is not uncommon for an analytical validation parameter to become more stringent as development progresses for the drug compound and its finished dosage form. The phase-appropriate approach for the process as well as the analytical development is summarized in figure 2.
Figure 2: Requirements for process and analytical method development using a phase-appropriate approach
The early development of the synthetic route of a drug substance and the corresponding analytical methods must be continuously documented according to the CMC Guidelines which form an important part of the evaluation of the toxicology studies, the IND submissions for the clinical phases, and finally the drug product approval [14].
Conclusion
Both the synthesis of a drug substance and its analytical methods should be developed and defined early on in a scientifically sound and data-based manner, as these are the foundation for meaningful toxicological evaluation and sufficient patient safety throughout the clinical phase.
Ardena applies phase-appropriate criteria for the qualification/validation of analytical methods during development. The key parameters taken into account are those referred to in ICH Q2 (R1): specificity, linearity, precision, accuracy, and LOD/LOQ. While the equipment used must be qualified from the outset, the full ICH validation of the process and analytical methods only takes place during the clinical development process. An efficient project plan combining drug substance synthesis and analytical method development through phase-appropriate deliverables along the development process ensures efficient pharmaceutical development and regulatory compliance.
References
Schlander et al (2021) How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. PharmacoEconomics 39:1243–1269
Wouters et al (2020) Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA ;323(9):844-853
Waring MJ et al (2015) An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nature Rev Drug Discov 14:475-486
Roberts RA et al (2014) Reducing attrition in drug development: smart loading preclinical safety assessment. Drug Discov Today 19(3):341-347
Brown DG & Boström J (2016) Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? Med. Chem. 2016, 59, 4443-4458