Enabling Complex Phase IIa Dosing Through Flexible Clinical Packaging Design
A shift to a four-arm, multi-dose Phase IIa protocol introduced new requirements for dosing accuracy, traceability, and patient usability.
Learn how Ardena designed a modular secondary packaging solution that integrated existing blister configurations and enabled seamless execution of the updated clinical design.
Achieving High Drug Tolerance in ADA Testing for a Long-Acting Monoclonal Antibody
Long-acting biologics present specific challenges for immunogenicity assessment, particularly when residual drug interferes with ADA detection. In this case study, Ardena outlines how a drug-tolerant ADA assay was established for a high-dose monoclonal antibody, enabling reliable detection in samples with high circulating drug levels.
Discover how targeted optimisation of a PandA-based approach delivered a robust, regulatory-ready solution for early clinical development.
Development of an assay to predict immunogenicity for an analogue of an endogenous protein
Case study
A highly promising analogue of an endogenous protein was identified as a drug candidate in preclinical studies in rats and monkeys. The sponsor decided to proceed with clinical studies in humans within 10 months, which was a major challenge as the data on immunogenicity required for the regulatory IND filing had to be generated. Due to this ambitious goal, the sponsor decided to partner with Ardena to develop a rational scientific immunogenicity assay according to the regulatory guidelines.
In the first step, an immunogenicity risk assessment was performed. As the compound was an analogue of an endogenous protein, the therapeutic protein was classified as a high-risk molecule for immunogenicity. This risk classification was based on the assumption that any antibodies generated and directed against the protein analogue may also be directed against the endogenous protein causing serious adverse reactions.
The most important basis for an immunogenicity assay is the development of a positive control and its production in sufficient quality and quantity to cover the assay development and immunogenicity screening. These positive control antidrug antibodies (ADA) are required for evaluation of the key parameters in the immunogenicity assay: assay sensitivity and drug tolerance. For this purpose, rabbits were immunized with the analogue protein to induce polyclonal antibodies against the drug. After 87 days, sufficient immune response was observed and the final bleed was harvested. After affinity purification of the final bleed, a quantity of approximately 35 mg of ADAs was obtained.
Case study
A highly promising analogue of an endogenous protein was identified as a drug candidate in preclinical studies in rats and monkeys. The sponsor decided to proceed with clinical studies in humans within 10 months, which was a major challenge as the data on immunogenicity required for the regulatory IND filing had to be generated. Due to this ambitious goal, the sponsor decided to partner with Ardena to develop a rational scientific immunogenicity assay according to the regulatory guidelines.
In the first step, an immunogenicity risk assessment was performed. As the compound was an analogue of an endogenous protein, the therapeutic protein was classified as a high-risk molecule for immunogenicity. This risk classification was based on the assumption that any antibodies generated and directed against the protein analogue may also be directed against the endogenous protein causing serious adverse reactions.
The most important basis for an immunogenicity assay is the development of a positive control and its production in sufficient quality and quantity to cover the assay development and immunogenicity screening. These positive control antidrug antibodies (ADA) are required for evaluation of the key parameters in the immunogenicity assay: assay sensitivity and drug tolerance. For this purpose, rabbits were immunized with the analogue protein to induce polyclonal antibodies against the drug. After 87 days, sufficient immune response was observed and the final bleed was harvested. After affinity purification of the final bleed, a quantity of approximately 35 mg of ADAs was obtained.
Meso Scale Discovery’s Multi Array® technology is a highly sensitive immunoassay system based on electrochemiluminescence (MSD-ECL), which was applied to develop and validate a bridging immunoassay in rat, monkey and human serum. Due to the universal nature of this assay for use in rat, monkey and human matrices, the assay development time and costs could be kept at a minimum.
Figure 1: Assay format, picture from MSD (Meso Scale Discovery)
In order to run the MSD-ECL, the analogue protein needed to be labelled with biotin and a sulfo-tag. Direct labelling of the drug of interest was not possible since it was formulated in an amine buffer. The problem was solved by a buffer exchange whereby the analogue protein was dialysed against a suitable buffer (PBS pH 7.4) before starting the labelling procedure. After dialysis, sufficient amounts of biotinylated drug as well as sulfo-tagged drug (at least 2 mg per label) were generated in the Ardena lab to cover a long period of immunogenicity development and screening. It is important to avoid lot-to-lot variabilities as much as possible as the labelled antibodies are considered to be critical reagents and will be required throughout the pre-clinical and clinical development. Nevertheless, Ardena is confident in its ability to reproduce the biotinylated and sulfo-tagged drug based on the internal procedures, assuring reproducibility in manufacturing of a new lot of labelled drug if required.
The assay development started with optimization of the conditions for the biotinylated and sulfo-tag labelled drug in the assay, followed by defining the matrix effect (minimal required dilution of the sample). The assay sensitivity was evaluated and drug tolerance determined. In this assay a mastermix approach was used, in which the sample containing ADAs was incubated for 2 hours at room temperature with a mastermix containing equal concentrations of biotinylated and sulfo-tag labelled drug (0.5 µg/mL of each labelled drug). The complex of biotinylated drug – ADA – sulfo-tag labelled drug was detected on a blocked MSD Gold Streptavidin plate.
A negative control pool was prepared per matrix by pooling from a sufficient number of individuals to cover a broad range of inter-individual variability. The negative controls consisted of at least 34 individual sources of rat and monkey serum and 51 individual sources of human serum. Positive control samples were prepared by the addition of different concentrations of the positive control antibody to the negative control pool.
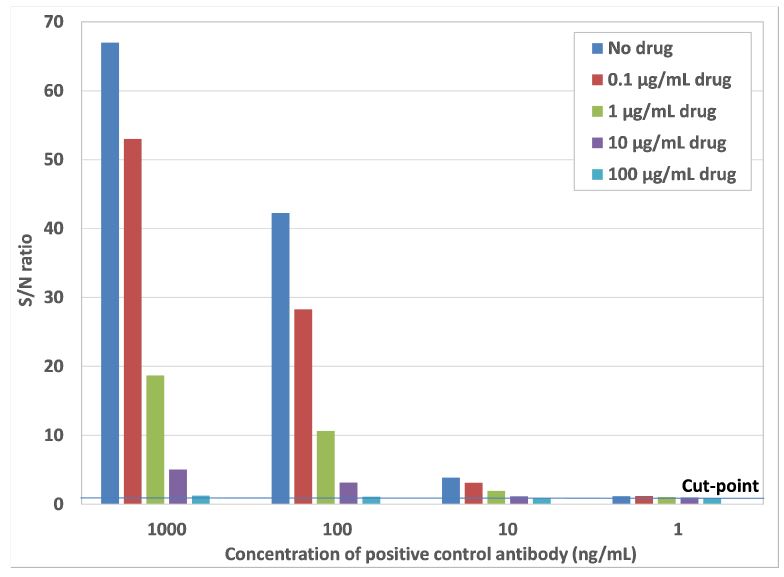
The minimal required dilution (MRD) was set at a 10-fold dilution of the samples containing ADAs, as this dilution gave a response close to the response of non-specific binding. The assay sensitivity and drug tolerance were very well within the acceptable range, as shown in the graph below. The method development results were discussed in detail with the sponsor before proceeding with validation of the immunogenicity assay. Based on the results and expert discussion it was decided that no pre-treatment of samples was required to optimize the drug tolerance. Furthermore the amount of excess drug to be used in the confirmation assay was set at 10 µg/mL, which resulted in complete inhibition of the response at low anti-drug antibody concentrations.
Figure 2: Graph from case study showing the assay sensitivity in the absence and presence of several concentrations of drug. The assay sensitivity is between 1 and 10 ng/mL anti-drug antibodies, which is well below the regulatory required sensitivity of 100 ng/mL. The drug tolerance is between 10 and 100 µg/mL of drug at an anti-drug antibody concentration of 100 ng/mL.
The immunogenicity assay was successfully validated under GLP according to a standard 3-tiered approach (i.e. screening, confirmation and titration assay) in rat and monkey serum. The validation in human serum is currently ongoing and the collaboration will continue throughout the IND filing and if successful, eventually progressing further into the clinical studies.
Conclusion
The development of an immunogenicity assay has become an integral part of biotherapeutic compounds that should begin at the earliest stages of drug development. Since there is no universal approach, the validity of the immunogenicity assay depends on expertise and the methodological approach used. Furthermore, valid immunogenicity assays should be considered throughout the clinical program and contribute to risk mitigation and patient safety. With its expertise and technological equipment in immunogenicity testing, Ardena can contribute at all stages of drug development.
Applying the science of advanced dissolution testing to accelerate product approval
Case study
Drug repurposing has become a rich source of safe and effective new therapeutic options against unmet medical needs. With the appearance of SARS CoV2 and the pandemic spread of severe Covid-19, drug repurposing has intensively being used to identify potential new drug candidates. One of the potential drug candidates was a small molecule prodrug with poor solubility in ethanol, practically insoluble in water and rapid hydrolyzation in aqueous media. The drug was marketed as an immediate release tablet for short term acute treatment. Since the drug showed efficacy against viruses including promising results to be effective against SARS CoV2 reformulation was required to achieve sufficient plasma level over the duration of 12 hours for a twice daily oral dosing regimen.
Case study
Drug repurposing has become a rich source of safe and effective new therapeutic options against unmet medical needs. With the appearance of SARS CoV2 and the pandemic spread of severe Covid-19, drug repurposing has intensively being used to identify potential new drug candidates [1]. One of the potential drug candidates was a small molecule prodrug with poor solubility in ethanol, practically insoluble in water and rapid hydrolyzation in aqueous media. The drug was marketed as an immediate release tablet for short term acute treatment. Since the drug showed efficacy against viruses including promising results to be effective against SARS CoV2 reformulation was required to achieve sufficient plasma level over the duration of 12 hours for a twice daily oral dosing regimen. Using available PK data from existing formulations (e.g. IR) combined with application of in-silico tools like PBBK modeling, predictions on formulation enhancements can be made [2]. For this compound it was predicted that an extended release formulation could lower the dose by ~ 40 % while maintaining a constant plasma levels in the therapeutic range. An extended release oral formulation was developed for the clinical trials that meet the release characteristics determined in the Target Product Profile for the new indication. Following successful phase 3 clinical trials of the extended release formulation, the FDA challenged the use of the dissolution test method derived from the immediate release formulation. Due to the BCS classification of the compound and the extended release properties of the formulation a dissolution method should serve as a surrogate for the in-vivo performance with sufficient discriminatory power. Consequently and following the pre-NDA meeting a discriminatory dissolution test had to be developed capable to demonstrate similarity between the extended release clinical formulation batches as well as providing an understanding of the critical quality attributes (CQA) and the critical process parameter (CPP) [3]. The ambitious time lines required the expertise and flexibility of Ardena’s resources to respond timely to the FDA requirements and finalize the NDA program as scheduled.
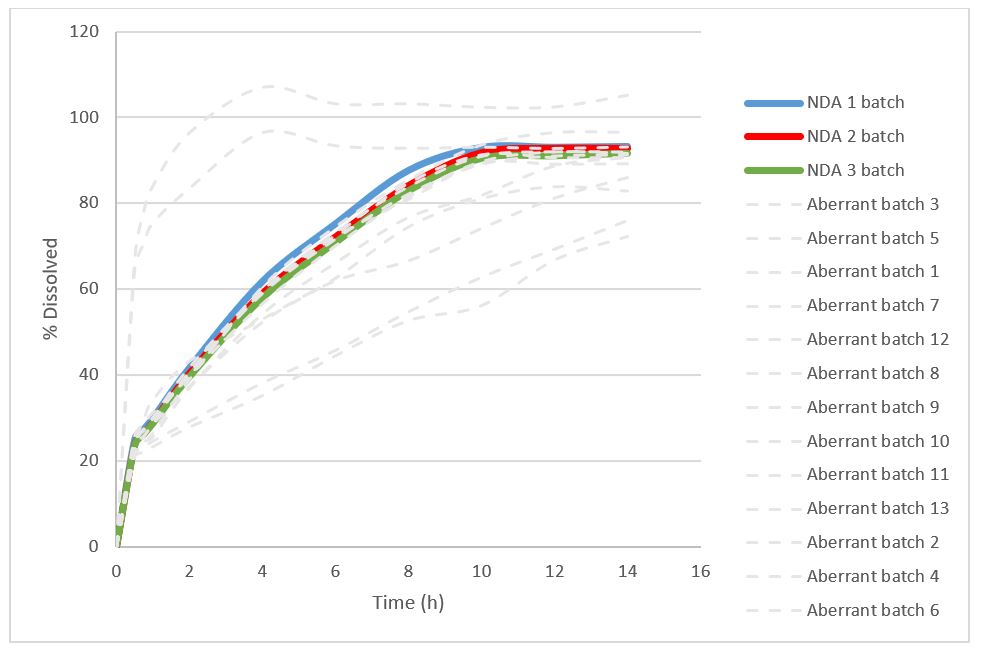
After reviewing the data and available samples from the immediate and extended release formulation Ardena defined a rational set of Design of Experiments. In a first step, a series of dissolution media, varying in pH and composition were performed using the USP type 2 (paddle) method at standard conditions for the immediate release as well as the newly developed extended release formulation. The dissolution screening included the quantification of the drug and its hydrolyzed derivative. The multi-media screening provided important dissolution patterns enabling the selection of two lead media compositions for the surfactant screening. After screening multiple surfactants a single surfactant was identified at two different concentrations to yield in the highest dissolution rate of the active and its degradation product. Additional comparative dissolution experiments were conducted on the available samples from the immediate and extended release formulation comparing the two media and surfactant levels which led to the selection of the most discriminatory composition. Finally, the conditions of the USP Type 2 method were modified to optimize the method by using different temperatures, vessel geometries and rotational speeds. To fulfill the regulatory requirements the method was validated according to the FDA guidelines on dissolution testing across the different product samples available. The final dissolution method than served to demonstrate the similarity between the clinical batches and provide the product as well as to serve process and product understanding for manufacturing and filing. Therefore, a risk assessment was performed and thirteen aberrant samples batches were manufactured to investigate the impact of the critical material attribute and process parameter variabilities. The comparative discriminatory dissolution data of this aberrant and the 3 registration batches are shown in figure 1. The dissolution method demonstrated the similarity of the clinical batches and identified the critical variabilities through non-similarity of the respective aberrant batch. Preferably representative aberrant batches should be produced on full scale equipment.
Figure 1: Dissolution profiles of aberrant core tablets and registration reference batches
The case study confirmed that advanced dissolution testing is an underestimated tool in accelerating drug product development into the clinics. The discriminatory dissolution test should be established early on in the program, ideally late phase 2 or early phase 3, to guide formulation development and clinical supplies as well as it is a tool to mitigate risk throughout the development, especially for drug repurposing or 505(b)(2) submissions [4]. Relying on Ardena’s experienced scientists provide fast and flexible resources for discriminatory dissolution test method development to support even the most ambitious development programs.
References
Wang & Guan (2021) COVID-19 drug repurposing: A review of computational screening methods, clinical trials, and protein interaction assays. Med Res Rev. 2021;41:5–28
Miller et al (2019) Physiologically based pharmacokinetic modelling for First-In-Human Predictions: An updated model building strategy illustrated with challenging industry case studies. Clin Pharmacokin 58:727–746
Yu et al (2014) Understanding pharmaceutical Quality-by-Design AAPS J 16(4):771-783
Freije et al (2020) Review of Drugs Approved via the 505(b)(2) Pathway: Uncovering drug development trends and regulatory requirements. Ther Innov 54(1):128-38
Discriminatory dissolution test method in late development
Case study: Building discriminatory power into dissolution testing in late development – a key milestone for the fast product approval
Dissolution testing was introduced into pharmaceutical practice after the occurrence of serious side effects of phenytoin after calcium sulfate was replaced by lactose. As a result, the FDA introduced the USP Dissolution type 1 (Basket method) test as a QC test in 1971 followed by USP Dissolution type 2 (Paddle method) test in 1978. Over the years, FDA developed further guidance to advance in-vitro dissolution testing into an important tool for development, quality control and drug approval. Statistical methods were implemented to compare the similarity or dissimilarity between two dissolution profiles, bio-relevant media were requested for predicting IVIVC and more specific guidelines were release for immediate and modified release products as well as the solubility and permeability characteristics of the drugs according to the BCS drug classification. With the growing knowledge on biopharmaceutics and predictive in-silico tools, dissolution testing has evolved into a key element for drug development and regulatory assessments. This includes that the dissolution test method has to include clinically relevant dissolution specification and must have proven discriminating power between variations outside the acceptance criteria.
Building discriminatory power into dissolution testing in late development – a key milestone for the fast product approval
Dissolution testing was introduced into pharmaceutical practice after the occurrence of serious side effects of phenytoin after calcium sulfate was replaced by lactose [1]. As a result, the FDA introduced the USP Dissolution type 1 (Basket method) test as a QC test in 1971 followed by USP Dissolution type 2 (Paddle method) test in 1978. Over the years, FDA developed further guidance to advance in-vitro dissolution testing into an important tool for development, quality control and drug approval. Statistical methods were implemented to compare the similarity or dissimilarity between two dissolution profiles, bio-relevant media were requested for predicting IVIVC and more specific guidelines were release for immediate and modified release products as well as the solubility and permeability characteristics of the drugs according to the BCS drug classification [2]. With the growing knowledge on biopharmaceutics and predictive in-silico tools, dissolution testing has evolved into a key element for drug development and regulatory assessments [3, 4]. This includes that the dissolution test method has to include clinically relevant dissolution specification [5] and must have proven discriminating power between variations outside the acceptance criteria [6].
Under increasing time pressure in the early development (e.g. phase 1) of new drugs, enhanced therapeutics (e.g. 505(b)(2)) or generics, the development of appropriate dissolution test methods is often limited to determine an adequate dissolution media, which provides sink conditions across the entire dose range of the product. Developing a predictive and discriminatory dissolution test method following the first in human trials has to be considered essential for rapid development of the pivotal clinical trial samples, commercial formulation, the regulatory filing and efficient commercial manufacturing. The earlier this important milestone is achieved in the development phase, the faster and more reliably approval and commercialization will occur [7].
Case study
A known small molecule with a water solubility > 1 mg/ml showed promising results to slow down the progression of multiple sclerosis at a dose of 100 mg tid. The product was formulated as an immediate release capsule. For the phase 1 clinical trial the standard USP type 2 (paddle) dissolution test method with a pH adjusted buffer was used to prove consistent dissolution between the development and the clinical batch. During pre-NDA discussions, the FDA challenged the existing dissolution test and requested evidence that the dissolution method chosen had sufficient discriminating power to assure that the product specification and hence critical clinical performance criteria are met.
Based on Ardena’s expertise a pragmatic, yet rational and systematic scientific approach was defined to develop the discriminatory dissolution method in due time. An assessment on the critical material attributes and critical process parameter of the product led to the manufacturing of 9 aberrant small batches serving for comparison to the clinical phase 1 batch as a reference. The available data from the existing dissolution method were analyzed to select potential dissolution media for the dissolution screen of the 10 different samples. The similarity factor (ƒ2) test was applied to the dissolution profiles of the batches to the reference batch at each time interval. The results demonstrated high sensitivity for one dissolution media discriminating between batches. . In a final design of experiments, the dissolution method was further fine-tuned by modifying the dissolution test procedural parameter (e.g. paddle speed) for the most discriminating power using again the similarity factor (ƒ2) test according to the FDA guidelines [7]. With the newly developed discriminating dissolution test method, 3 of the aberrant batches were found outside the similarity specification providing important understanding on the product and process parameter required to consistently meet the quality criteria of the product.
Development of discriminative dissolution method during phase 3 results in 3-6 months time win
While the project passed all IND phases successfully without any further delay, the discriminatory dissolution test revealed important formulation factors like composition, excipients grades and processing affecting the dissolution of the active. This confirms that a discriminating dissolution method available in late phase 2 or early phase 3 of the project, can accelerate the NDA process by 3 to 6 months . Partnering with Ardena’s expertise and scientific resources covering the entire development cycle into the clinics and beyond can mitigate the risk to reach the clinical proof of concept and regulatory approval on schedule.
References
Tyrer et al (1970) Outbreak of anticonvulsant intoxication in an Australian city. Med. J. 4 (5730), 271–273
Amidon et al (2006) A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation in Vitro Drug Product Dissolution and in Vivo Pharm Res, 12 (3), 413–420
EMA (2012) Guideline on quality of oral modified release products EMA/492713/2012
FDA (2017) Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. Guidance for Industry
Heimbach et al (2019) Dissolution and Translational Modeling Strategies Toward Establishing an In Vitro-In Vivo Link—a Workshop Summary Report. AAPS J 21:29
Gray (2018) Power of the Dissolution Test in Distinguishing a Change in Dosage Form Critical Quality Attributes. AAPS PharmSciTech 19(8): 3328.3332
McAllister et al (2020) Developing Clinically Relevant Dissolution Specifications for Oral Drug Products—Industrial and Regulatory Perspectives. Pharmaceutics 12: 19 (2020)].
FDA (1997) Dissolution Testing of Immediate Release Solid Oral Dosage Forms; Guidance for Industry
Juan Jose Serrano
Your molecule deserves the right expert.
Our scientists have spent careers solving the formulation problems that stop other CDMOs. Let's talk about yours.